Genes & Cancer
Loss of TRP53 (p53) accelerates tumorigenesis and changes the tumor spectrum of SJL/J mice
Jane A. Branca1, Benjamin E. Low1, Ruth L. Saxl1, Jennifer K. Sargent1, Rosalinda A. Doty1, Michael V. Wiles1, Beth L. Dumont1 and Muneer G. Hasham1
1 The Jackson Laboratory, Bar Harbor, Maine, USA
Correspondence to: Muneer G. Hasham, email: [email protected]
Keywords: TRP53, p53, SJL/J, tumorigenesis
Received: November 07, 2019
Accepted: January 06, 2020
Published: January 21, 2020
Copyright: © 2020 Branca et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ABSTRACT
Known as the guardian of the genome, transformation-related protein 53 (TRP53) is a well -known tumor suppressor. Here, we describe a novel TRP53 deficient mouse model on a tumor prone background—SJL/J mice. The absence of TRP53 (TRP53 nullizygosity) leads to a shift in the tumor spectrum from a non-Hodgkin’s-like disease to thymic lymphomas and testicular teratomas at a very rapid tumor onset averaging ~12 weeks of age. In haplotype studies, comparing tumor prone versus tumor resistant Trp53 null mouse strains, we found that other tumor suppressor, DNA repair and/or immune system genes modulate tumor incidence in TRP53 null strains, suggesting that even a strong tumor suppressor such as TRP53 is modulated by genetic background. Due to their rapid development of tumors, the SJL/J TRP53 null mice generated here can be used as an efficient chemotherapy or immunotherapy screening mouse model.
INTRODUCTION
The product of the transformation-related protein (Trp53) is one of the most researched tumor suppressors in biomedical research, with currently more than 97,000 research/review articles published to date. Known as the guardian of the genome, the function of TRP53 protein (TP53, or, in humans, p53) has been well studied, and its functions include transcriptional regulation, DNA repair, cell cycle check-point control, apoptosis, autophagy and senescence (for review, see [1-9]). The lack of a functional p53 gene product in humans leads to Li-Fraumeni syndrome predisposing the patient to a spectrum of early-onset cancers (for review, see [10-12]). Importantly, it has been shown that since p53 in humans is located at a distal end of chromosome 17 (17p13.1), the loss of this piece of chromosome, and thereby p53, generally accelerates cancers [3, 13].
Since TRP53 has many crucial functions in biological systems, a number of mouse models have been made in order to elucidate TRP53 function more fully [14-18]. Of principal note here, Trp53 mutations have been shown to synergize with loss-of-function mutations in other tumor suppressor genes generally accelerating tumor development and progression. For example, p53 deficiency synergizes with: Rb deficiency in a conditional mouse model to produce metastatic prostate cancer [19]; NUP98 translocation in a NUP98-HOXD13– driven mouse model to accelerate complications of myelodysplastic syndrome [20]; and mutations in Apc to promote mammary neoplasia [21]. Hence, the disrupting of TRP53 has become a tool to accelerate the growth of tumors that develop from mutations in other tumor suppressor genes allowing more rapid and efficient study of these tumors.
Swiss Jim Lambert (SJL/J) mice, developed from three different sources of Swiss Webster mice, have become widely used owing to their high incidence of reticulum cell sarcomas. They develop lymphomas within their first year [22-31] that resemble Hodgkin’s disease [23-25] as well as B-cell non-Hodgkin’s lymphomas [26-31]. In the presence of IL-21, the tumors arising from SJL/J mice resemble human angioimmunoblastic T-cell lymphoma [22]. In addition to cancer models, these mice have been used as models for experimental autoimmune encephalomyelitis (EAE) [32], aggression [33], spontaneous myopathy in limb girdle muscular dystrophy [34], and cardiovascular disease, due to their resistance to developing atherosclerotic aortic lesions even on high fat diet [35]. The SJL/J strain is highly susceptible to mouse adenovirus 1, making it also a model for infectious disease studies [36]. Therefore, the SJL/J strain of mice is a highly valued disease model to test therapeutics for a diversity of conditions and diseases.
We previously used these mice to test to what extent the chemotherapeutic 2-deoxy-D-glucose (2DG) can alleviate the tumor burden of SJL/J mice exhibiting terminal stages of cancer [37]. 2DG is a structural analog to glucose and blocks glycolysis leading to intracellular ATP depletion, sensitizing tumor cells to radiation therapy and chemotherapy [38]. However, 2DG at high doses show hypoglycemic and adverse cardiac effects, and at tolerable doses fail to show a significant antitumor effect in many in vivo experiments in both mice and humans [39]. Our 2DG studies in SJL/J mice were primarily conducted to test to what extent this toxic chemotherapeutic could have reduced adverse effects when combined with other compounds. At doses that did not elicit adverse effects, 2DG alone given to mice with a SJL/J background was able to significantly shrink tumors [37]. However, the tumors develop resistance to 2DG after four weeks, after which the tumor growth re-emerges [37]. Although the penetrance of spontaneous tumorigenesis in SJL/J is >95%, the time it takes to develop such tumors is ~one year, with a range between nine months to 1.2 years [24]. This protracted pathogenesis means that it is difficult to generate sufficient cohorts of mice to be tested in a timely fashion. Therefore, to address this challenge, we hypothesized that removing Trp53 would accelerate tumor development, and thereby enable the ability to test the efficacy of novel combinations of chemotherapies or immunotherapies within a more operationally convenient timespan.
Here, we generate a TRP53 null SJL/J mice using CRISPR Cas9 with two guide RNAs aimed at deleting exon 4 of the Trp53 gene. Upon analysis, TRP53 was expressed in +/+ mice, reduced in heterozygous mice, and absent in -/- mice. Homozygous null mice showed a significantly shorter time of onset of tumorigenesis, and a reduced survival, with tumors being detected as early as 11 weeks of age, faster than all the other models commonly available. However, we also found that the ablation of TRP53 in SJL/J mice shifted the tumor spectrum to thymic lymphomas, testicular teratomas and rhabdomyosarcomas rather than the typical Hodgkin’s/non-Hodgkin’s lymphomas that SJL/J wild type mice develop. This unexpected shift in tumor spectrum to thymic lymphomas is observed with TRP53 mutations in other strains such as C57BL6/J, 129S1, BALB/c, FVB/NJ, and C3H/J, suggesting that deregulation of TRP53 leads to specific types of cancers depending upon the genetic background [14-18]. We further interrogated the haplotypes of these strains, comparing tumor-prone TRP53 null strains versus the non-tumor-prone strains, and found that a majority of the alleles that modulate the tumor incidence rate have either cancer, immune system or DNA-repair related functions. The immune system and DNA repair systems are known to be critical in regulating tumorgenesis. This result suggests that not even a strong tumor suppressor gene such as Trp53 is canalized, and the genetic background will modify the resulting neoplasticity.
RESULTS
Generation of Trp53-/- SJL/J mice
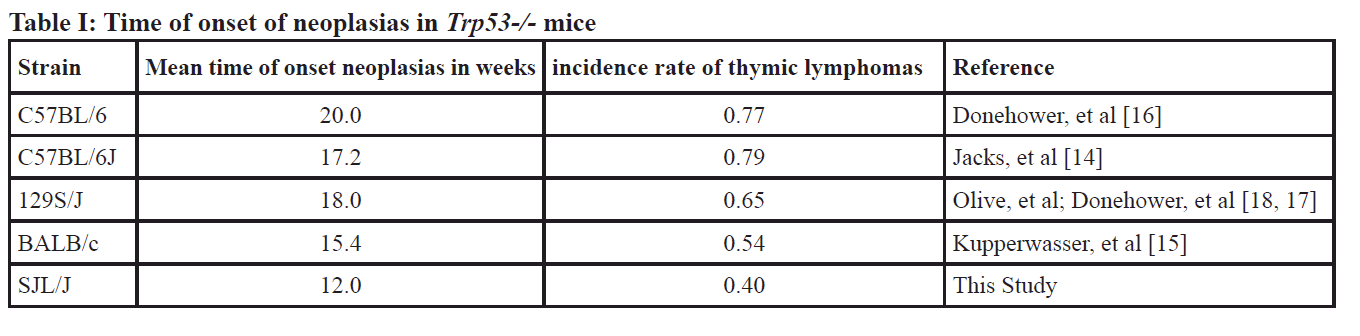
Based on the ENSEMBL database (Release 98), exon 3 of TRP53 is the first exon that is sufficiently large enough to generate guides that can target the gene. Furthermore, functional domains of TRP53 are further downstream of this exon, thus targeting this exon to introduce a frame shift mutation is anticipated to lead to a null mutation. According to the ENSEMBL database, Trp53 transcripts contain 5’UTR sequences that include ATG sequences that could be putative translational start sites. Thus, herein exon 3 is designated as exon 4, assuming that upstream starts sites yield an additional exon.
To generate p53 knockout alleles in the SJL/J mouse strain, superovulated female mice were mated and zygotes were collected for microinjection. Of the 28 resulting offspring, 15 (54%) had modifications at one or both target sites; six had deletions of the intervening sequence between each guide and thus considered “dropouts.” Founder dropout candidates were crossed with wild type SJL/J mice and N1 heterozygotes were genotyped to characterize the mutant alleles. One allele was ultimately selected to establish a new mutant mouse strain (Figure 1A, 1B), designated SJL/J-Trp53<em2Mvw>/Mvw JAX stock 33940 (i.e. a 176bp deletion; ∆176). Genotyping and characterization of mutant alleles was accomplished using PCR (FWD: 5’- TCCAGACTTCCTCCAGAAGATA, REV:5’-CCTCTGTGCTTGGCTTCA) and Sanger sequencing. The wild type PCR product is 624bp and the ∆176 mutant allele results in a 448bp amplicon (Figure 1C).
The mutant allele was maintained in heterozygous mice. Heterozygous mice were mated to obtain the null, heterozygous or wild type alleles. Breeding outcomes showed that the allele frequency was below expected Mendelian ratios for the homozygotes at 1:3:1=wild type:heterozygous:null alleles (data from 10 litters, with litter sizes of ~ 5 mice). However, all mice (i.e., null, heterozygous, wild type) survive through weaning at a frequency similar to that of the parental SJL/J strain.
Detection of TRP53 protein
From the nucleotide sequences, it is predicted that the ∆176 allele should yield a stop codon after 69 residues (predicted 7.8kDa), leaving the transactivation domain intact but deleting all domains downstream (Figure 1D). Intact TRP53 is predicted to be 42.5kD, however, the protein appears around 53kDa [5]. The livers, the largest and least affected organ, from a homozygous null, heterozygous and wild type littermate from a heterozygous ∆176/+ mating pair were isolated and lysates probed via western blot with a monoclonal antibody raised against serine 20 of human TRP53, which cross reacts with mouse TRP53. Western analysis showed two bands of proteins ~50kD (Figure 1E) as observed previously [5]. Our western blot observation and analysis of the ∆176 allele showed no detectable protein, suggesting that the protein might be degraded since the antibody raised against serine 20 should have been able to detect the truncated protein. The heterozygotes for this allele produce less WT TRP53 than that observed in the wild type littermates (β-tubulin was used as a loading control (Figure 1E)). In order to confirm the observed absence of the protein in the ∆176 mutant, we performed an ELISA using antibodies directed towards the N-terminus to confirm the absence of the protein in mutant mice and to quantify the amount of protein in wild-type and heterozygous mice (Figure 1F). The ELISA confirmed the western blot results, revealing that the amount of TRP53 in the livers (10uL of lysate) was 1929 pg/mL in +/+, 422 pg/mL in the +/- and 13 pg/mL in the -/-. Considering the standard error of detection in the -/- mice was 88 pg/mL, the amount in the null can be assumed to be zero.
Survival of Trp53-/- and Trp53+/- mice
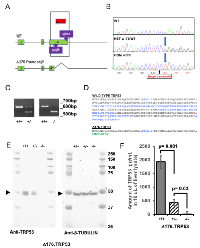
Mice carrying the ∆176 allele were further characterized for tumor spectrum and survival. The absence of TRP53 significantly (P = 0.02) shortens the lifespan of the mice from ~one year to a median of 12 weeks (Figure 2). At 35 weeks, all of the homozygous null mice (N = 5) and four of 14 heterozygous mice had died, but none of the wild type mice (N = 6) had died. Three of the 10 heterozygous mice that lived showed signs of tumorigenesis. The reduction in survival has been observed before in at least five other strains of mice with TRP53 mutations, suggesting that regardless of the genetic background, the loss of TRP53 reduces survival [14-16, 18].
The SJL/J ∆176.Trp53-/- mice have shown a faster development of neoplasticity than the tumor prone model BALB/c, or tumor resistant models, such as C57BL/6 (Table I) which suggests that the genetic background can modulate the phenotype even of a strong tumor suppressor such as TRP53.
Tumor spectrum of the TRP53 null mice
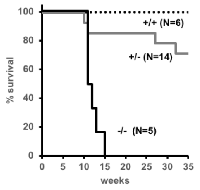
No tumors were detected or observed in the wild type littermates of the mice at 35 weeks of age. TRP53 null (-/-) SJL/J mice developed thymic lymphomas (40%), testicular teratomas (40%) or were found dead without any visible tumors (20%) (Figure 3A). The heterozygous (+/-) mice developed sarcomas (15%) or were found dead without tumors, and therefore, non-cancer related cause of death (7%) (Figure 3A); none developed thymic lymphomas or testicular teratomas. There was no gender bias in the development of thymic lymphomas, and only half of male SJL/J ∆176.Trp53-/- mice developed testicular teratomas. A shift to thymic lymphomas is consistent with the effects of Trp53 nullizygosity in other strains [14, 16-18],
Upon closer examination of the major types of tumors derived from +/- and -/- mice, the following was observed (Figure 3B-3E): The thymic lymphomas exhibited canonical neoplastic round cells (Figure 3B) with a high mitotic rate (red arrowheads) of >20 mitotic figures per high powered field (hpf). Necrosis was minimal, but there were numerous apoptotic cells (white circles) throughout the mass. The mice also showed variable splenomegaly due to infiltration by small clusters of neoplastic lymphocytes. These features are typical of Trp53-/- tumors observed in previous models [14, 16].
The testicular tumor masses which were isolated from -/- mice were classical teratomas, comprised of neoplastic tissues from all three germ layers. Accordingly, the tumors were composed of a mixture of cell types (Figure 3C): ectoderm (neuropil, red asterisks), mesoderm (bone, arrow; cartilage, arrowhead; adipose tissue and muscle), and endoderm (squamous to columnar ciliated epithelium with goblet cells consistent with respiratory epithelium, black asterisks). The neoplastic cells in all of the teratomas had a high mitotic rate (>20 mitotic figures/hpf), and also showed areas of necrosis and/or hemorrhage.
The third type of tumor observed in +/- mice but not in -/- SJL/J mice were rhabdomyosarcomas (Figures 3D). These tumors were composed of neoplastic cells that were primarily spindle cells, with a smaller proportion of round cells, as well as some multinucleated cells (arrows) and strap cells (asterisks). The neoplastic cells have a high mitotic rate (>10 mitotic figures/hpf) and mild necrosis was seen in the tumor. The spleens were large due to plasmacytosis (asterisks), and the red pulp have a marked increase in megakaryocytes (arrowheads) (Figure 3E).
In summary, TRP deficiency in a SJL/J background shifts the cancer spectrum from primarily Hodgkin’s and non-Hodgkin’s lymphoma to thymic lymphomas, testicular teratoma and rhabdomyosarcomas.
Incidence and time-of-onset of thymic lymphomas
As compared to refractive strains such as C57BL6/J or 129S, which show >65% incidence rate for thymic lymphomas, fewer SJL/J mice developed thymic lymphomas (40%), matching more closely to the tumor prone strain BALB/c (54%) (Table I). However, thymic lymphomas in the refractive strains took longer to develop than in the cancer-prone strains. For example, the average time-of-onset of thymic lymphomas exceeded 17 weeks in C57BL6/J or 129S mice, but was only 12 weeks in SJL/J mice (Table 1). Thus, tumor prone strains have a lower propensity to develop thymic lymphomas, but develop them much faster than the more refractive strains. This suggests that the genetic background may play a role in modulating the effect of Trp53 nullizygosity. It also suggests Trp53 knockout in SJL/J mice offer a more operationally convenient timespan for the study of thymic lymphomas.
Genomic analysis of strain haplotype sharing nominates putative candidate modifiers of Trp53-/- mice
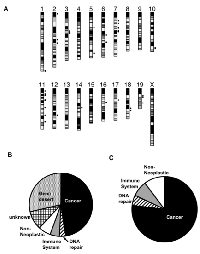
The commonly used laboratory mouse strains are descended from a small founder pool of individuals and thus only capture a limited amount of genetic diversity. Across more than 97% of the mouse genome, the genetic variation present in classical laboratory strains can be reconciled into fewer than 10 distinct haplotypes. Thus, we reasoned that putative modifiers of the strain-specific Trp53-/- tumor spectrum may reside in genomic regions where the refractive C57BL/6J and 129S1/SvImJ strains carry the same haplotype, but where the susceptible SJL/J and BALB/cJ strains both carry distinct haplotypes. Included in the tumor resistant phenotype group are also FVB/NJ and C3H/J, which according to JAX repository data, develop a similar tumorigenic phenotype as the C57BL6/J mice (JAX Stock # 002899 and #002547, unpublished data). Using the known haplotype patterns that are shared or distinct among strains, we identified distinct regions that could be associated with putative tumor spectrum modifiers. Overall, only 0.207% of the mouse genome (~5.45Mb) exhibited this expected haplotype profile across these six strains (Figure 4A). We grouped these haplotypes together based on the genes in which they harbored into six distinct categories, including haplotypes that harbored at least one known (i) cancer gene, (ii) DNA repair gene, (iii) immune system gene, (iv) non-neoplastic gene, (v) genes with unknown function and (vi) gene deserts (Figure 4B). After removing gene deserts and unknown function genes, we reanalyzed these data (Figure 4C). The pie charts generated show that the majority of the genes in these potential modifier regions are known to be directly associated with cancer, or function in DNA repair or the immune system, which are known to influence and modify tumorigenesis (Supplementary Figure I). Several compelling candidate genes reside in these regions, including genes Egfr, Rad51b, and Foxp1, which all have been shown to modify p53 tumorigenesis, as discussed below. Therefore, we have identified the likely/putative/probable genomic regions harboring Trp53 modifiers that make strains such as BALB/cJ and SJL/J tumor prone.
DISCUSSION
While the absence of functional TRP53 (also known as p53 in humans, TP53 in mouse) in humans causes Li Fraumeni syndrome, the loss of the distal end of chromosome 17p13.1 has been linked to a wide spectrum of early-onset cancers and acceleration of tumor growth [13]. In mice, the loss of TRP53 activity is used to generate unique mouse models for studies in transcriptional regulation, cell cycle regulation, apoptosis, and genome instability [1-3, 5-9, 13-21]. In addition, TRP53 has been mutated to rescue embryonic lethality. For example, to study non-homologous end joining in mouse models, the removal of the Trp53 gene rescues the embryonic lethal DNA repair protein Ligase IV [43]. Of relevance to this study, TRP53 nullizygosty was successfully used to accelerate tumorigenesis in a tumor prone mouse. This is a unique model that can be used for rapid cancer and therapeutic studies. In addition, by adding this model to the library of current TRP53 mutated models, the genetic haplotypes that modify such as a strong tumor suppressor can be evaluated.
Based on this evidence, we used CRISPR-Cas9 to delete part of exon 4 (or exon 3 depending on the functional ATG site, see Figure 1A) on the TRP53 gene, with the goal that this would generate a TRP53 knock-out, since most of the domains for TRP53 function lie downstream or are a part of this exon. Such a genetic modification of this gene would lead to an acceleration of tumorigenesis and facilitate the testing of novel combinations of chemotherapeutics. As predicted, Trp53 nullizygosity has accelerated the pace of tumor development leading to a decrease in survival. The speed with which these tumors appear is remarkably fast - 12 weeks on average, as compared to other models (Table 1). Therefore, this model is useful to study rapid tumorigenesis in mouse models.
We also showed that a decrease (null) of TRP53 changed the tumor spectrum from a non-Hogdkin’s like disease to thymic lymphoma in SJL/J mice, similar to that observed in other strains such as seen in Table I. When polymorphisms are used to classify similar mice, BALB/cJ, C57BL/6J and 129S/J are quite diverse from SJL strains [44, 45]. Yet, thymic lymphomas appear in all of these strains. The data presented here in Table I exemplify canalization, which is defined as a measure of the ability of a population to produce a similar phenotype regardless of variability of its environment or genotype. The lack of fully functional TRP53 is a powerful driver of canalization, that regardless of the genotype leads to a single major phenotype (i.e., thymic lymphomas).
However, there is a caveat to this conclusion. In the context of Trp53 nullizygosity, the tumor prone strains SJL/J and BALB/c show the lowest incidence of thymic lymphomas but they develop much faster as compared to C57BL6/J and 129S/J, which show the highest frequency of thymic lymphomas, but they develop much slower (Table I). These results lead us to speculate that the link between Trp53 nullizygosity and tumorigenesis may not be as simple as the removal of a checkpoint in the cell cycle, ablation of apoptosis or any one simple step in a pathway, but a more complex interplay between different genomes and the propensity to develop cancer.
Based on the notion that the genome can change the phenotype of an assumed strong canalized gene such as Trp53, we surveyed the literature and JAX repository data, and analyzed the genomic differences based on haplotypes between tumor prone strains and tumor resistant strains. Here we have identified a number of likely/putative/probable genomic regions harboring Trp53 modifiers that make strains such as BALB/cJ and SJL/J tumor prone. Genetic and functional tests will be required to evaluate whether and how these putative candidates may modulate the landscape of tumor formation in the absence of Trp53-/-. The function of specific genes such as Egfr, Rad51b, and Foxp1 have been shown to be modulated by Trp53 [46-48]. A specific example is that mutations in p53 amplify EGFR family signaling to promote mammary tumorigenesis [46]. RAD51B is a homologous recombination DNA repair protein that interacts with TRP53 [49, 50], and its activity influences cell cycle checkpoint control, independent of its role in homologous recombination in breast cancer [51]. It is also known that RAD51B, being a homologous recombination protein, shares its function to maintain genome stability with TRP53 [52, 53]. FOXP1 is a transcription factor essential for the development of major organs and known to be a tumor suppressor in prostrate and breast cancers [54, 55]. In fact, Foxp1 directly represses p53-dependent regulatory proteins in neoplastic B-cells, suggesting a strong role in immune modulation [56]. As we currently understand normal anti-cancer responses, a functional immune system is a key component that suppresses cancer [57-59]: Foxp1 controls mature B-cell survival and development, and is a regulator for CD4+ T cells [60, 61]. Thus, it is also likely that Foxp1 is an essential component that controls the lymphoid immune system, and thereby a modulator of tumorigenesis. In all of these cases, there are direct or indirect mechanisms that could be employed by these genes to modulate Trp53-dependent tumorigenesis.
Interestingly, known gene products of Trp53 modifiers, such as MDM2, MDM4 and CDKN2A (p14(ARF)), did not appear in our analysis [2]. It is possible that the aforementioned genes are modifiers of Trp53 mutations resulting in different tumor spectrums, rather than a complete loss or gain of TRP53 that changes the tumor incidence, which was the analysis done herein.
Genetically Engineered Mouse Models (GEMMS) are key to testing novel chemotherapy and immunotherapy compounds [62]. However, tumors may take more than several months to develop. To shorten tumor latency time, Trp53 is often deleted [63, 64]. Also, the arising tumors from a TRP53 null mouse can be directly tested for drug efficacy studies (for summary see: [62]). This mouse can be used as a model to test T-cell malignancies, testicular teratomas and rhabdomyosarcomas, with the advantage that it is faster than any other available model.
The concept of whether genetic background influenced the tumor incidence or spectrum of Trp53-/- mice was tested decades ago: Donehower et al (1995) studied Trp53-/- and +/- mice and their wild-type littermates from either 129Sv or mixed C57BL/6 × 129/Sv. The 129/Sv mice showed accelerated tumorigenesis (Table I) compared with p53-deficient counterparts of C57BL/6 × 129/Sv genetic background, the 129/Sv mice resemble the C57BL/6 strainwith respect to tumor onset (Table I). This suggests that the genetic background indeedmodulates the TRP53 effect on tumor incidence. We have compared multiple studies (Table I), and show that different genetic backgrounds can remarkably influence tumor onset.
This study did not analyze the tumor spectrum of heterozygous mice. In addition, we reported data up to 35 weeks when a quarter of the heterozygous mice showed tumors or were found dead. Heterozygous mice express less than half the amount of TRP53 in their tissues as compared to wild type tissues. It is understood that Trp53+/- cells have the propensity to develop loss of heterozygosity (LOH) depending on the type of cell. These two aforementioned phenomena, the lowered amount of TRP53 and LOH, potentially drive the tumor onset and spectrum in the heterozygous mice [5, 6, 53]. Dissecting the two mechanisms (dose of TRP53 and LOH) that would contribute to a heterozygous cell’s propensity to become neoplastic in this tumor prone mouse model is a subject of future study.
Here, we present a mouse model of Trp53 inactivation in the tumor prone background SJL/J. This genetic modification of Trp53 leads to rapid tumorigenesis as compared to current models. Further, apart from the common thymic lymphomas and rhabdomyosarcomas, they also exhibit testicular teratomas. These models can now join the pantheon of Trp53 mutations that can be used to further dissect the role of TRP53 in the context of tumor-prone models versus tumor-resistant models, and reveal genetic and epigenetic cancer etiologies.
MATERIALS AND METHODS
Mice
SJL/J mice used in this study were obtained from, and bred and housed at The Jackson Laboratory (Bar Harbor, Maine). Mice were provided food and water ad libitum and were housed on a 12-hour light, 12-hour dark cycle. All procedures were approved by The Jackson Laboratory Institutional Animal Care and Use Committee (IACUC).
CRISPR mediated knock out of Trp53
Approximately 2-5 picoliters of Cas9 mRNA (at 100 ng/ul, Trilink), Cas9 protein (at 30 ng/ul, PNABio), and two sgRNAs (at 50 ng/ul each) were delivered by microinjection into the pronucleus of SJL/J zygotes as previously described [40]. Truncated guides (TRU-sgRNAs) [41] were designed with the assistance of online software, Breaking Cas [42], in order to minimize off-target cutting. The guides were designed to target exon 4 of the Trp53 gene (target sites: 5’-GAGCTCCTGACACTCGGA and 5’- GCCAAGTCTGTTATGTGCA) and were made using a HiScribe kit (New England Biolabs). Microinjected zygotes were transferred to pseudopregnant females, brought to term and screened at two to three weeks of age by PCR flanking exon 4. These founder mice were backcrossed with SJL/J, and resulting mice heterozygous for the mutation were then inbred to homozygosity.
Survival criteria
Mice were monitored daily from the time of birth and diagnosed with a tumor as soon as a visible abnormal growth appeared. The tumors were allowed to grow until the mice showed signs of illness and required euthanasia which then was recorded as the date of death. The need for euthanasia was independently verified by our Clinical and Laboratory Animal Medicine personnel. In addition, mice that were found dead were necropsied and diagnosed for the cause of death. All mice were terminated at 35 weeks of age.
Western blotting
Snap frozen tissues were pulverized with a Spectrum Bessman Tissue Homogenizer (Fisher Scientific # 08-418-3) on dry ice. A portion of each tissue was transferred into a pre-weighed 1.5mL tube and the weight of each tissue sample was determined. The tissue samples were subsequently suspended in 100uL of chilled RIPA (Millipore-Sigma # R0278) with complete mini protease inhibitors (Roche #11836153001) and PhosSTOP phosphatase inhibitor (Roche #4906845001) per 40ug of tissue and homogenized on ice for 10 s with individual sterile pestles (Fisher Scientific #12-141-364). After 5 min on ice, homogenization was repeated. After incubating on ice for a further 30 min with a brief vortex every 5 min, the samples were spun at 10,000 × g for 10 min at 4°C. The supernatants were decanted and their protein concentration determined by the Bradford Assay (Bio-Rad # 500-0205) using BSA (Bio-Rad # 5000206) for the standard curve. Gel samples were prepared and denatured at 95°C for 5 min. 50 ug total protein per tissue was separated on 10% Bis-Tris Protein Gels (Fisher Scientific #NP0302BOX) with MOPS SDS Running Buffer (Fisher Scientific #NP0001) in duplicate. The gels were transferred to nitrocellulose (Fisher Scientific #IB3010-32) using an iBlot Gel Transfer Device (Fisher Scientific). The blots were blocked in 3% BSA in 25mM Tris·Cl, 2.7mM KCl, 137mM NaCl and 0.03% TWEEN-20 (TBST) at room temperature for 1 h. One blot was then placed in p53 (1C12) monoclonal antibody (Cell Signaling Technology #2524) diluted 1:2000 in 3% BSA-TBST and the second blot was placed in 3% BSA-TBST with a β-tubulin antibody (Millipore-Sigma #T4026) diluted 1:10,000. Both blots were agitated on a shaker at 4° C overnight. The blots were washed with TBST for 15min and then with 5 min washes then placed in 3% BSA-TBST with goat anti-Mouse IgG (H + L)-HRP conjugate (Bio-Rad # 170-6516) diluted 1:20,000 for 1 h at room temperature. After washing with TBST for 15min and then with 5 min washes, the blots were developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Fisher Scientific # PI-34087) and imaged with a G-box (Syngene).
ELISA for TRP53 quantification
Samples used for western blotting (see above) were quantified using the Abcam Mouse p53 ELISA Kit (Abcam # ab224878).
Statistics and protein prediction:
For survival curves, P-values were calculated by log-rank (Mantel-Cox) test using Prism7 v7.0d software. Protein sizes were predicted from the Protein Calculator (v3.4) (http://protcalc.sourceforge.net/)
Genome queries
Genome-wide haplotype strain distribution patterns were downloaded from the Mouse Phylogeny Viewer (http://msub.csbio.unc.edu/). A custom Perl script was used to identify genomic regions harboring a single shared haplotype between C57BL/6J, FVB/NJ, C3H/HeJ and 129S1/SvImJ, a second distinct haplotype in SJL/J, and a third haplotype in BALB/cJ. These genomic regions were then intersected with genes in the mm10 RefSeq database. We performed a manual inspection of the resulting gene list to nominate putative modifiers of Trp53 that could underlie observed strain differences in the tumor spectrum.
ACKNOWLEDGMENTS
We thank Byrant Perkins, Caitlin Lewis, and Cindy Avery for mouse care and genotyping. Susanne Sattler and Stephen Krasinski for advice and manuscript review.
We gratefully acknowledge the contribution of Microinjection, JAX Sanger Sequencing, Histopathological Sciences and Microscopy Service at The Jackson Laboratory for expert assistance with the work described in this publication.
Research reported in this publication was supported by The Linda Tallen and David Kane Educational Foundation and partially by the National Cancer Institute of the National Institutes of Health under Award Number P30CA034196, OD210259-0119 and The Jackson Laboratory. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTERESTS
None.
- 1. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018; 25:104–13. https://doi.org/10.1038/cdd.2017.169. [PubMed].
- 2. Basu S, Murphy ME. Genetic Modifiers of the p53 Pathway. Cold Spring Harb Perspect Med. 2016; 6:a026302. https://doi.org/10.1101/cshperspect.a026302. [PubMed].
- 3. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017; 170:1062–78. https://doi.org/10.1016/j.cell.2017.08.028. [PubMed].
- 4. Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016; 37:8471–86. https://doi.org/10.1007/s13277-016-5035-9. [PubMed].
- 5. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997; 88:323–31. https://doi.org/10.1016/s0092-8674(00)81871-1. [PubMed].
- 6. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009; 9:749–58. https://doi.org/10.1038/nrc2723. [PubMed].
- 7. Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018; 25:133–43. https://doi.org/10.1038/cdd.2017.174. [PubMed].
- 8. White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016; 6:a026120. https://doi.org/10.1101/cshperspect.a026120. [PubMed].
- 9. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016; 6:a026070. https://doi.org/10.1101/cshperspect.a026070. [PubMed].
- 10. Correa H. Li-Fraumeni Syndrome. J Pediatr Genet. 2016; 5:84–88. https://doi.org/10.1055/s-0036-1579759. [PubMed].
- 11. Vogel WH. Li-Fraumeni Syndrome. J Adv Pract Oncol. 2017; 8:742–46. [PubMed].
- 12. Valdez JM, Nichols KE, Kesserwan C. Li-Fraumeni syndrome: a paradigm for the understanding of hereditary cancer predisposition. Br J Haematol. 2017; 176:539–52. https://doi.org/10.1111/bjh.14461. [PubMed].
- 13. Seifert H, Mohr B, Thiede C, Oelschlägel U, Schäkel U, Illmer T, Soucek S, Ehninger G, Schaich M, and Study Alliance Leukemia (SAL). The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009; 23:656–63. https://doi.org/10.1038/leu.2008.375. [PubMed].
- 14. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994; 4:1–7. https://doi.org/10.1016/s0960-9822(00)00002-6. [PubMed].
- 15. Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol. 2000; 157:2151–59. https://doi.org/10.1016/S0002-9440(10)64853-5. [PubMed].
- 16. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215–21. https://doi.org/10.1038/356215a0. [PubMed].
- 17. Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA Jr, Park SH, Thompson T, Ford RJ, Bradley A. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 1995; 14:16–22. https://doi.org/10.1002/mc.2940140105. [PubMed].
- 18. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004; 119:847–60. https://doi.org/10.1016/j.cell.2004.11.004. [PubMed].
- 19. Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, Nikitin AY. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006; 66:7889–98. https://doi.org/10.1158/0008-5472.CAN-06-0486. [PubMed].
- 20. Xu H, Menendez S, Schlegelberger B, Bae N, Aplan PD, Gring G, Deblasio TR, Nimer SD. Loss of p53 accelerates the complications of myelodysplastic syndrome in a NUP98-HOXD13-driven mouse model. Blood. 2012; 120:3089–97. https://doi.org/10.1182/blood-2012-01-405332. [PubMed].
- 21. Méniel V, Hay T, Douglas-Jones A, Sansom OJ, Clarke AR. Mutations in Apc and p53 synergize to promote mammary neoplasia. Cancer Res. 2005; 65:410–16. [PubMed].
- 22. Jain S, Chen J, Nicolae A, Wang H, Shin DM, Adkins EB, Sproule TJ, Leeth CM, Sakai T, Kovalchuk AL, Raffeld M, Ward JM, Rehg JE, et al. IL-21-driven neoplasms in SJL mice mimic some key features of human angioimmunoblastic T-cell lymphoma. Am J Pathol. 2015; 185:3102–14. https://doi.org/10.1016/j.ajpath.2015.07.021. [PubMed].
- 23. Haran-Ghera N, Kotler M, Meshorer A. Studies on leukemia development in the SJL/J strain of mice. J Natl Cancer Inst. 1967; 39:653–61. [PubMed].
- 24. Kumar RK. Hodgkin’s disease. SJL/J murine lymphoma. Am J Pathol. 1983; 110:393–96. [PubMed].
- 25. Ouyang S, Han G, Wu X, Jiang Z, Feng JM. Accelerated progression of Hodgkin’s-like lymphomas in golli deficient SJL mice. Cell Immunol. 2016; 302:41–49. https://doi.org/10.1016/j.cellimm.2016.01.006. [PubMed].
- 26. Stavnezer J, Lasky JL, Ponzio NM, Scheid MP, Thorbecke GJ. Reticulum cell sarcomas of SJL mice have rearranged immunoglobulin heavy and light chain genes. Eur J Immunol. 1989; 19:1063–69. https://doi.org/10.1002/eji.1830190616. [PubMed].
- 27. Tang JC, Ho FC, Chan AC, Srivastava G. Clonality of lymphomas at multiple sites in SJL mice. Lab Invest. 1998; 78:205–12. [PubMed].
- 28. Lacoste-Collin L, Jozan S, Cances-Lauwers V, Pipy B, Gasset G, Caratero C, Courtade-Saïdi M. Effect of continuous irradiation with a very low dose of gamma rays on life span and the immune system in SJL mice prone to B-cell lymphoma. Radiat Res. 2007; 168:725–32. https://doi.org/10.1667/RR1007.1. [PubMed].
- 29. Thrush GR, Butch AW, Lerman SP. CD8 suppressor cell activity and its effect on CD4 helper cell-dependent growth of SJL/J B-cell lymphomas. Cell Immunol. 1989; 122:555–62. https://doi.org/10.1016/0008-8749(89)90101-9. [PubMed].
- 30. Sopchak L, King SR, Miller DA, Gabra N, Thrush GR, Lerman SP. Progression of transplanted SJL/J lymphomas attributed to a single aggressive H-2Ds-negative lymphoma. Cancer Res. 1989; 49:665–71. [PubMed].
- 31. Thrush GR, Placey JL, Valeriote FA, Lerman SP. The CD4 cell dependency of SJL/J B-cell lymphomas as a target for cyclophosphamide therapy. Cancer Commun. 1989; 1:301–10. https://doi.org/10.3727/095535489820874896. [PubMed].
- 32. Plinger B, Krishnamoorthy G, Berer K, Lassmann H, Bl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, Wekerle H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic Tcells recruit endogenous MOG-specific B cells. J Exp Med. 2009; 206:1303–16. https://doi.org/10.1084/jem.20090299. [PubMed].
- 33. Miner LL, Elmer GI, Pieper JO, Marley RJ. Aggression modulates genetic influences on morphine analgesia as assessed using a classical mendelian cross analysis. Psychopharmacology (Berl). 1993; 111:17–22. https://doi.org/10.1007/BF02257401. [PubMed].
- 34. Weller AH, Magliato SA, Bell KP, Rosenberg NL. Spontaneous myopathy in the SJL/J mouse: pathology and strength loss. Muscle Nerve. 1997; 20:72–82. https://doi.org/10.1002/(sici)1097-4598(199701)20:1<72::aid-mus10>3.0.co;2-3. [PubMed].
- 35. Paigen B. Genetics of responsiveness to high-fat and high-cholesterol diets in the mouse. Am J Clin Nutr. 1995; 62:458S–62S. https://doi.org/10.1093/ajcn/62.2.458S. [PubMed].
- 36. Spindler KR, Fang L, Moore ML, Hirsch GN, Brown CC, Kajon A. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J Virol. 2001; 75:12039–46. https://doi.org/10.1128/JVI.75.24.12039-12046.2001. [PubMed].
- 37. Wilson JJ, Chow KH, Labrie NJ, Branca JA, Sproule TJ, Perkins BR, Wolf EE, Costa M, Stafford G, Rosales C, Mills KD, Roopenian DC, Hasham MG. Enhancing the efficacy of glycolytic blockade in cancer cells via RAD51 inhibition. Cancer Biol Ther. 2019; 20:169–82. https://doi.org/10.1080/15384047.2018.1507666. [PubMed].
- 38. Dwarakanath BS, Singh D, Banerji AK, Sarin R, Venkataramana NK, Jalali R, Vishwanath PN, Mohanti BK, Tripathi RP, Kalia VK, Jain V. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J Cancer Res Ther. 2009 (Suppl 1); 5:S21–26. https://doi.org/10.4103/0973-1482.55136. [PubMed].
- 39. Vijayaraghavan R, Kumar D, Dube SN, Singh R, Pandey KS, Bag BC, Kaushik MP, Sekhar K, Dwarakanath BS, Ravindranath T. Acute toxicity and cardio-respiratory effects of 2-deoxy-D-glucose: a promising radio sensitiser. Biomed Environ Sci. 2006; 19:96–103. [PubMed].
- 40. Low BE, Kutny PM, Wiles MV. Simple, Efficient CRISPRCas9-Mediated Gene Editing in Mice: strategies and Methods. Methods Mol Biol. 2016; 1438:19–53. https://doi.org/10.1007/978-1-4939-3661-8_2. [PubMed].
- 41. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014; 32:279–84. https://doi.org/10.1038/nbt.2808. [PubMed].
- 42. Oliveros JC, Franch M, Tabas-Madrid D, San-Le D, Montoliu L, Cubas P, Pazos F. Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBLgenomes. Nucleic Acids Res. 2016; 44:W26771. https://doi.org/10.1093/nar/gkw407. [PubMed].
- 43. Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000; 5:993–1002. https://doi.org/10.1016/s1097-2765(00)80264-6. [PubMed].
- 44. Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, Sargent EE, Asquith S, Crew V, Johnson KA, Robinson P, Scott VE, Wiles MV. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004; 14:1806–11. https://doi.org/10.1101/gr.2825804. [PubMed].
- 45. Petkov PM, Cassell MA, Sargent EE, Donnelly CJ, Robinson P, Crew V, Asquith S, Haar RV, Wiles MV. Development of a SNP genotyping panel for genetic monitoring of the laboratory mouse. Genomics. 2004; 83:902–11. https://doi.org/10.1016/j.ygeno.2003.11.007. [PubMed].
- 46. Yallowitz AR, Li D, Lobko A, Mott D, Nemajerova A, Marchenko N. Mutant p53 Amplifies Epidermal Growth Factor Receptor Family Signaling to Promote Mammary Tumorigenesis. Mol Cancer Res. 2015; 13:743–54. https://doi.org/10.1158/1541-7786.MCR-14-0360. [PubMed].
- 47. Aggarwal C, Davis CW, Mick R, Thompson JC, Ahmed S, Jeffries S, Bagley S, Gabriel P, Evans TL, Bauml JM, Ciunci C, Alley E, Morrissette JJ, et al. Influence of TP53 Mutation on Survival in Patients With Advanced EGFR-Mutant Non-Small-Cell Lung Cancer. JCO Precis Oncol. 2018; 2018:1–29. https://doi.org/10.1200/PO.18.00107. [PubMed].
- 48. Karnam S, Kottu R, Chowhan AK, Bodepati PC. Expression of p53 & epidermal growth factor receptor in glioblastoma. Indian J Med Res. 2017; 146:738–45. https://doi.org/10.4103/ijmr.IJMR_1179_15. [PubMed].
- 49. Shu Z, Smith S, Wang L, Rice MC, Kmiec EB. Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53(-/-) background. Mol Cell Biol. 1999; 19:8686–93. https://doi.org/10.1128/mcb.19.12.8686. [PubMed].
- 50. Wu M, Sheng Z, Jiang L, Liu Z, Bi Y, Shen Y. Overexpression of RAD51B predicts a preferable prognosis for non-small cell lung cancer patients. Oncotarget. 2017; 8:91471–80. https://doi.org/10.18632/oncotarget.20676. [PubMed].
- 51. Lee PS, Fang J, Jessop L, Myers T, Raj P, Hu N, Wang C, Taylor PR, Wang J, Khan J, Jasin M, Chanock SJ. RAD51B Activity and Cell Cycle Regulation in Response to DNA Damage in Breast Cancer Cell Lines. Breast Cancer (Auckl). 2014; 8:135–44. https://doi.org/10.4137/BCBCR.S17766. [PubMed].
- 52. Sullivan MR, Bernstein KA. RAD-ical New Insights into RAD51 Regulation. Genes (Basel). 2018; 9:E629. https://doi.org/10.3390/genes9120629. [PubMed].
- 53. Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006; 13:1003–16. https://doi.org/10.1038/sj.cdd.4401903. [PubMed].
- 54. Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Takahashi S, Homma Y, Urano T, Aburatani H, Inoue S. Integrative analysis of FOXP1 function reveals a tumor-suppressive effect in prostate cancer. Mol Endocrinol. 2014; 28:2012– 24. https://doi.org/10.1210/me.2014-1171. [PubMed].
- 55. Koon HB, Ippolito GC, Banham AH, Tucker PW. FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets. 2007; 11:955–65. https://doi.org/10.1517/14728222.11.7.955. [PubMed].
- 56. van Keimpema M, Grüneberg LJ, Mokry M, van Boxtel R, Koster J, Coffer PJ, Pals ST, Spaargaren M. FOXP1 directly represses transcription of proapoptotic genes and cooperates with NF-κB to promote survival of human B cells. Blood. 2014; 124:3431–40. https://doi.org/10.1182/blood-2014-01-553412. [PubMed].
- 57. Janssen LM, Ramsay EE, Logsdon CD, Overwijk WW. The immune system in cancer metastasis: friend or foe? J Immunother Cancer. 2017; 5:79. https://doi.org/10.1186/s40425-017-0283-9. [PubMed].
- 58. Candeias SM, Gaipl US. The Immune System in Cancer Prevention, Development and Therapy. Anticancer Agents Med Chem. 2016; 16:101–07. https://doi.org/10.2174/1871520615666150824153523. [PubMed].
- 59. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018; 32:1267–84. https://doi.org/10.1101/gad.314617.118. [PubMed].
- 60. Patzelt T, Keppler SJ, Gorka O, Thoene S, Wartewig T, Reth M, Fster I, Lang R, Buchner M, Ruland J. Foxp1 controls mature B cell survival and the development of follicular and B-1 B cells. Proc Natl Acad Sci USA. 2018; 115:3120–25. https://doi.org/10.1073/pnas.1711335115. [PubMed].
- 61. Garaud S, Roufosse F, De Silva P, Gu-Trantien C, Lodewyckx JN, Duvillier H, Dedeurwaerder S, Bizet M, Defrance M, Fuks F, Bex F, Willard-Gallo K. FOXP1 is a regulator of quiescence in healthy human CD4+ T cells and is constitutively repressed in T cells from patients with lymphoproliferative disorders. Eur J Immunol. 2017; 47:168–79. https://doi.org/10.1002/eji.201646373. [PubMed].
- 62. Kim WY, Sharpless NE. Drug efficacy testing in mice. Curr Top Microbiol Immunol. 2012; 355:19–38. https://doi.org/10.1007/82_2011_160. [PubMed].
- 63. Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007; 448:807–10. https://doi.org/10.1038/nature06030. [PubMed].
- 64. Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007; 447:966–71. https://doi.org/10.1038/nature05886. [PubMed].
Last Modified: 2020-06-10 01:04:12 EDT
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.