Genes & Cancer

SPARC overexpression alters microRNA expression profiles involved in tumor progression
Bhavesh K. Ahir1, Nasya M. Elias1 and Sajani S. Lakka1
1 Section of Hematology and Oncology, Department of Medicine, University of Illinois College of Medicine at Chicago, Chicago, IL, USA
Correspondence to: Sajani S. Lakka, email: [email protected]
Keywords: Medulloblastoma, SPARC, microRNA profiling, Tumor progression, Oncomine
Received: December 05, 2016
Accepted: February 11, 2017
Published: February 14, 2017
ABSTRACT
Medulloblastoma is the most common malignant brain tumor in children. SPARC (secreted protein acidic and rich in cysteine), a multicellular non-structural glycoprotein is known to be involved in multiple processes in various cancers. Previously, we reported that SPARC expression significantly impairs medulloblastoma tumor growth in vitro and in vivo and also alters chemo sensitivity. MicroRNAs are a class of post-transcriptional gene regulators with critical functions in tumor progression. In addition, microRNA (miRNA) expression changes are also involved in chemo-resistance. Herein, we assessed microRNA (miRNA) profiling to identify the functional network and biological pathways altered in SPARC-overexpressed medulloblastoma cells. A total of 27 differentially expressed miRNAs were identified between the control and SPARC-overexpressed samples. Potential messenger RNA (mRNA) targets of the differentially expressed miRNA were identified using Ingenuity Pathway Analysis (IPA). Network-based functional analyses were performed on the available human protein interaction and miRNA-gene association data to highlight versatile miRNAs among the significantly deregulated miRNAs using the IPA, and the biological pathway analysis using the PANTHER web-based tool. We have identified six miRNAs (miR-125b1*, miR-146a-5p, miR-181a-5p, miR-204-5p, miR-219-5p and miR-509-3p) that are associated with SPARC sensitivity by comparison of miRNA expression patterns from the SPARC treated cells with the control cells. Furthermore, pathway enrichment analysis outline that these six microRNAs mainly belong to biological processes related to cancer related signaling pathways. Collectively, these studies have the potential to indicate novel biomarkers for treatment response and can also be applied to develop novel therapeutic treatment for medulloblastoma.
INTRODUCTION
Medulloblastoma (MB) is the most common malignant brain tumor diagnosed in children that account for approximately 16-20 % of all pediatric brain tumors arising in the cerebellum [1-3]. Despite improved clinical outcomes with multimodal treatment including surgery, radiation and chemotherapy, tumor reappearance is frequent, and a 5-year survival rate of these patients is 60-70 % [4-7]. Furthermore, children diagnosed with medulloblastoma often suffer from cognitive and physical dysfunction resulting from current treatment of resection, followed by the long term toxicities associated with radiation and chemotherapy [8]. In addition, they have a tendency to disseminate throughout the central nervous system and identifying new molecular targets are required to improve outcomes.
SPARC also called osteonectin or basement membrane (BM 40) is a 32–35 kDa multifunctional collagen or calcium-binding ECM glycoprotein located at 5q33.1 and it consists of a single polypeptide with 285 amino acids including three biological structural domains; the acidic N-terminal (NT) domain, a follistatin-like domain and a Ca2+ binding extracellular domain [9-11]. It belongs to the matricellular family of secreted non-structural glycoproteins [12, 13]. In human carcinogenesis, SPARC plays remarkable roles in multiple biological functions including cell proliferation, cell cycle, adhesion, motility, apoptosis, extracellular (ECM) remodeling and on the activity of metalloproteases [14, 15]. Moreover, SPARC is also thought to play a critical role in differentiation of several cell types including epithelial differentiation [16], parietal endoderm differentiation, cardiomyogenesis in embryoid bodies [17], and differentiation of myoblasts [18]. SPARC plays an important role in neural and/or glial differentiation [19]. We have previously shown that SPARC expression caused neuronal differentiation [20] and caused autophagy mediated cell death [21]. Our studies also showed that SPARC enhances radiotherapy response in medulloblastoma in vitro and in vivo [22]. Understanding the role of signaling pathways involved in surviving and inducing cell death with SPARC expression is important for the development of more effective tumor therapies including SPARC gene therapy alone or in combination with other drugs [20, 23]. Several research groups have analyzed the mRNA expression profile of brain tumors to reveal novel gene markers for diagnosis and therapy and to better understand the regulatory pathways and genetic networks involved in medulloblastoma [24, 25].
MicroRNAs (miRNAs), an abundant class of ~22-nucleotide non-coding RNAs, regulate the expression of genes at post transcriptional level [26]. It has become increasingly evident that miRNA plays an essential role in the brain including neurogenesis and differentiation [27, 28]. To date, miRNAs are thought to regulate almost 60 % of all protein-coding genes in humans and participate in the regulation of almost every cellular processes investigated [29]. Recent studies have shown that approximately 60-70 % of the currently identified miRNAs are expressed in the brain [30, 31]. The miRNA-related genetic alterations are implicated in multiple human diseases and pathological processes including cancer [32]. MicroRNAs not only act as oncogenes by inhibiting translation of tumor suppressor mRNAs, but they can also act as tumor suppressor genes as well, by inhibiting translation of oncogenic mRNAs [26, 33, 34]. Several groups of miRNAs have been identified to regulate the expression of tumor-associated genes [35, 36], while others seem to hold prognostic value in predicting the survival of cancer patients [37].
In this study, we identified the miRNA expression profile in SPARC overexpressed human medulloblastoma cells using microarray technology. Using bioinformatics tools and systems biology approach, we systematically identified differentially expressed miRNAs, miRNA-mRNA predictive targets, biological functions and disease signature, network analysis and the biological signaling pathways of gene targeted by these SPARC modulated differentially expressed miRNAs. We further selectively confirmed six miRNAs with statistically significant differential expression by miRNA quantitative real-time polymerase chain reaction (qRT-PCR) in SPARC expressed cells and identified that the expression of these six miRNA targets are altered in human medulloblastoma patient samples.
RESULTS
SPARC expression in medulloblastoma cells
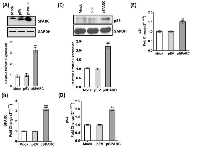
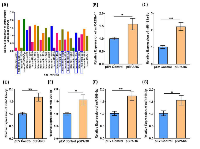
We previously demonstrated that human medulloblastoma tissue samples expressed very low or minimal levels of SPARC when compared with normal cerebellum [20]. Furthermore, we also showed that SPARC protein and mRNA levels were increased up to 3–4-folds in D425 and UW228 medulloblastoma cell lines transfected at a 2 μg/ml of pSPARC compared to controls [23]. Herein, we sub-cloned human SPARC full-length cDNA into a pcDNA3.1 mammalian expression vector (pSPARC). We transfected medulloblastoma D283 cells with pSPARC for 36 h. Total protein and total RNA was isolated from the cells at 36 h after post-transfection, and immune blot analysis and qRT-PCR was performed to detect SPARC expression levels in medulloblastoma cells. We found more than 3-fold increase in relative SPARC protein and mRNA transcript expression in D283 medulloblastoma cells as compared to parental (mock) and empty vector (pEV) controls (p < 0.01; Figure 1A and 1B). Our previous studies also indicated that the Sub-G1 region corresponding to apoptotic cells was increased by 56.7 ± 4% in SPARC overexpressed D283 cells as compared to 5.0 ± 2% in empty vector treated controls. In. addition, SPARC significantly increased TUNEL-positive apoptotic cells in D283 medulloblastoma cells in a concentration dependent manner. Further, we also demonstrated that SPARC overexpression increased caspase-3 and caspase-8 activities and enhanced PARP cleavage when compared to mock or an empty vector controls in D283 medulloblastoma cells [20, 21]. Fluorescence activated cell sorting (FACS) analysis indicated that SPARC expressed cells were arrested in G2/M phase compared to mock or empty vector treated controls in medulloblastoma cells [23]. We therefore explored potential mechanisms of the altered cell cycle arrest profile. We determined p53 and p21 level in SPARC overexpressed cells by immunoblotting and qRT-PCR analysis. SPARC expression induced p53 and p21 levels by 2–3 fold compared to controls (Figure 1C, D and E). These studies suggest that p53/p21(WAF1/CIP1) pathway is a critical component of a cell cycle regulatory pathway(s) that controls the occurrence of G2M arrest in SPARC expressed D283 medulloblastoma cells.
Medulloblastoma miRNA profiles identified putative candidate miRNA markers
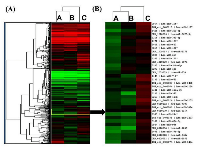
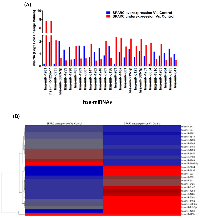
We used custom multi-species microarrays containing 1209 probes covering 1220 human mature miRNAs, to detect differentially expressed miRNAs in SPARC overexpressed D283 medulloblastoma cells. The custom microarray chip containing 1209 probes covering 1220 human miRNAs present in the miRBase version 16.0 database released in 2010 [38]. Figure 2A shows the unsupervised hierarchical clustering of the 729 miRNAs with acceptable detection intensities. A very remarkably different clustering pattern was observed in SPARC overexpressed versus SPARC under expressed medulloblastoma samples compared to parental/control medulloblastoma samples (Figure 2A and B).
To compile a set of targeted miRNAs for further investigation and confirmation, the list of 623 Log2 transformed and normalized raw spot intensities were examined for difference between the group by at least 2-fold differences between the control and SPARC overexpression and/or SPARC underexpression (Supplementary Material, Table S1). A total of 27 differentially expressed probes exhibited at-least two-fold difference between the control and SPARC overexpression or SPARC-underexpression samples (Figure 3A). Hierarchical unsupervised clustering of medulloblastoma using the most significantly differentially expressed miRNAs is represented in a heat map (Figure 3B). A set of 27 differentially expressed miRNAs have been clustered by unsupervised hierarchical clustering analysis, generating a dendogram that shows a clear separation of SPARC overexpression versus control and SPARC underexpression versus control (Figure 3B).
Prediction of transcriptional targets of differentially expressed miRNAs using IPA
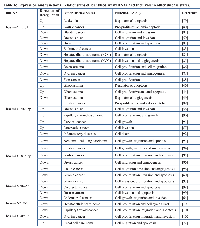
We performed in silico analysis to understand genomic changes likely regulated by this set of 27 differentially expressed miRNAs. Using the Ingenuity miRNA Target Filter, based on knowledgebase of predicted and experimentally observed miRNA-mRNA relationships, we identified 1,076 mRNA that were experimentally or predicted targets of the 15 miRNAs. Inclusion of only the experimentally observed downstream mRNA targets resulted in a set of 6 miRNAs with 82 target mRNA, of which 81 were unique (Table 2; Supplementary Material, Table S2). This in silico analysis predicted miR-125b-1* to target 3 mRNAs, miR-146a-5p to target 47 mRNAs, miR-181a-5p to target 14 mRNAs, miR-204-5p to target 12 mRNAs, miR-219a-5p to target 5 mRNAs, and miR-509-3p to target 1 mRNAs for a total 82 targets (see Table 2).
Network and functional enrichment of SPARC overexpression mediated miRNA targets
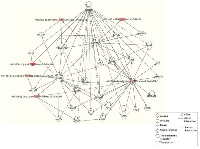
Functional enrichment analysis of microRNAs and their target genes was performed to identify unique, similar and common sets of genes/proteins. We constructed molecular networks with the 81 unique mRNAs predicted to be regulated by 6 differentially expressed miRNAs in SPARC expressed medulloblastoma cells using the IPA. Fig. 4B indicates that the most significantly enriched network were associated with inflammatory cytokines (p < 10-10) in SPARC over expressed cells. Tumor necrosis factor (TNF) was identified as a key node in these interactions. The top most 10 significantly associated networks were inflammatory disease, inflammatory response to immune cell, cellular movement and immune cell trafficking, cellular development including cellular growth and proliferation, cell morphology, hematological system development and function, cell-to-cell signaling, cell cycle and inflammatory response to neurological disease (Table 3; Supplementary Material, Table S3).
Pathway analysis of differentially expressed miRNAs
We performed pathway analysis using PANTHER database in order to understand which signaling pathways are altered with SPARC expression in medulloblastoma cells. The 81 unique mRNA gene list was entered into the PANTHER database in order to identify significantly over- and under-represented pathways, molecular functions and biological processes, in comparison with a reference gene (Homo sapiens Whole Genome list) (Table 4). From the initial list of 81 unique genes, PANTHER was unable to map 3 mRNAs. For this reason, all analyses were performed on a total 78 gene targets. Table 4 shows statistically significant (p < 0.05) pathways, molecular function classes and biological process relevant to the 78 gene targets genes. In all cases, a statistically significant (p < 0.05) overrepresentation of the number of target genes per category was also observed. A total of 6 biological signaling pathways were significantly associated with SPARC mediated expression in medulloblastoma cells (p < 0.05) (Table 4). The top significant pathways associated with SPARC mediated expression in medulloblastoma cells include toll receptor signaling pathway, interleukin signaling pathway, apoptosis signaling pathway, angiogenesis signaling pathway, inflammation mediated chemokine and cytokine signaling pathway and platelet derived growth factor (PDGF) signaling pathway.
Analysis of putative microRNA targets in medulloblastoma patient samples
To further investigate the biological relevance of the six miRNAs differentially regulated in SPARC expressed cells, we looked for the known targets of these six miRNAs in medulloblastoma patient samples (Table 5). The analysis was focused on the research of targets experimentally validated in previously published studies using publicly available human medulloblastoma datasets within the Oncomine Database (www.oncomine.org/) [39, 40]. We mined 81 mRNA targets of the six differentially expressed miRNAs in desmoplastic medulloblastoma cancer type samples versus normal control samples (n=4 normal brain cerebellum control samples and n=14 desmoplastic patient samples). In all cases, a statistically significant altered expression of the number of target genes was observed in medulloblastoma patient samples (see Table 5 and Supplementary Material, Table S2).
Expression of SPARC modulated miRNAs in medulloblastoma
Relative changes in miRNA expression observed in microarray analysis were validated using qRT-PCR. We evaluated the 6 miRNAs identified based on in silico prediction for experimentally observed miRNA-mRNA target predictions using the IPA in SPARC overexpressed cells (Figure 5A). qRT-PCR was performed on miR-125b-1* (FC = 1.57), miR-181a-5p (FC = 2.21), miR-146a-5p (FC = 1.68), miR-204-5p (FC = 1.57), miR-509-3p (FC = 1.73) and miR-219-5p (FC = 1.57) in SPARC overexpressed medulloblastoma cells (pSPARC) compared to empty vehicle (pEV) treated control samples. There was a significant increase in relative fold change of all the six microRNAs (Figure 5B-G). We next evaluated the role of these six microRNAs in other disease states along with their functional relationships as summarized in Table 6. The top significant pathways associated with these microRNAs that are regulated by these six microRNAs include cell proliferation, growth, invasion, angiogenesis and apoptosis.
DISCUSSION
MicroRNA signature can predict the prognosis and therapy response for appropriate therapeutic management. Our previous studies have shown that SPARC expression was very low in human medulloblastoma tissue samples when compared with normal cerebellum tissue samples [20]. We also showed that SPARC expression inhibits medulloblastoma tumor growth in an intracranial mouse model [21, 23]. We also observed that SPARC expression induced the expression of neuronal marker genes (such as MAP-2, NeuN, nestin and neurofilament) [20]. Further, we also showed that SPARC expression enhanced radio response and combined treatment of SPARC and irradiation resulted in increased cell death when compared to cells treated with irradiation alone in vitro and in vivo. SPARC expression suppressed irradiation-induced checkpoints-1,-2 and p53 phosphorylation and DNA repair gene, XRCC1. In addition, SPARC expression suppressed irradiation induced SOX-4 mediated DNA repair [22]. Further, we also showed that SPARC altered cisplatin sensitivity by modulating the Let-7f-1 miRNA/HMGB1 axis in medulloblastoma cells. In this study, we demonstrated that autophagy was involved in SPARC expression mediated resistance to cisplatin. Further, SPARC expression suppressed miR-let-7f-1 expression which resulted in disrupted repression of High Mobility Group Box 1 (HMGB1), a critical regulator of autophagy [41]. MicroRNAs can act as oncogenes or tumor suppressors and are capable of modulating several targets in multiple genetic pathways. Identification of a specific pattern of expression of miRNAs in SPARC expressed cells could shed light on the underlying mechanisms of SPARC mediated tumor suppressive and chemosensitising effects.We therefore performed global expression analysis for microRNAs using miRNA microarray to explore the miRNAome in SPARC expressed medulloblastoma and control cells. Twenty-seven miRNAs were identified as differentially expressed between SPARC expressed cells compared to control cells. In silico analysis using IPA analysis based on the research of targets experimentally validated in previously published studies indicated that miR-125b-1*, miR-146a-5p, miR-181a-5p, miR-204-5p, miR-509-3p and miR-219-5p were upregulated in SPARC expressed cells. The interactive networks and hubs revealed functional cooperatively of these six miRNAs and target groups in the various signaling pathways. The functional and pathway enrichment analysis identified that different oncogenic pathways regulated by these six microRNAs were previously demonstrated to be involved in cancer progression.
Our studies show that miR-125b was increased with SPARC expression. This is consistent with a recent study in which they showed that miR-125b was upregulated in medulloblastoma [42]. miR-125b is the most abundant miRNA in the brain and its play an important role in embryonic brain development including neural development by repressing multiple targets [43]. Furthermore, miR-125b on one hand promotes proliferation and growth of glial and neuroblastoma tumor cells in vivo and in vitro [44-46] and on the other hand, it behaves as a tumor suppressor in glioblastoma-associated endothelial cells, glioma stem cells and medulloblastoma [47-49]. Several studies also indicate that miR-125b functions as a tumor suppressor. The expression of miR-125b is downregulated in various human cancers including glioblastoma, prostate cancer, ovarian cancer, and breast cancer by suppressing oncogenes such as EST1, ERBB2, ERBB3, and BAK1 as its targets [50, 51]. Another study also demonstrated that silencing of miR-125b1 leads to the activation of the ETS1 proto-oncogene resulting in worse prognosis in breast cancer patients [51]. Further miR-125b was implicated in neuronal differentiation of mouse P19 embryonal carcinoma cells [52]. The depletion of miR-125b suppressed the proliferation of differentiated human neuroblastoma cells in vitro [53]. This is consistent with our previous studies demonstrating that SPARC expression suppressed proliferation and induced neuronal differentiation in medulloblastoma cells [20, 23] suggesting that mir-125b could be a key player in SPARC mediated tumor suppressive effects in medulloblastoma cells.Our data also indicates that SPARC expression enhanced miR-146a expression. This result is consistent with a demonstrated role of miR-146a as a tumor suppressor in several studies. Upregulation of miR-146a plays an inhibitory role in restricting the formation of glioma stem-like cells and tumor burden [54]. Overexpression of miR-146a inhibits the proliferation and survival of breast, prostate, and pancreatic cancer cells through the downregulation of its targets including ROCK1, EGFR, and MTA-2 [55-58]. Deletion of miR-146a spontaneously developed subcutaneous flank tumors in mice [59]. Ectopic expression of miR-146a inhibits tumor development of a human glioblastoma cell line in an orthotropic xenograft model by downregulation of Notch1, which plays a key role in neural stem cell maintenance and is a direct target of miR-146a [54]. Conversely, knockdown of miR-146a by microRNA sponge upregulates Notch1 and promotes tumorigenesis of malignant astrocytes [54]. We have previously demonstrated that SPARC stimulates neuronal differentiation of medulloblastoma cells by suppressing Notch signaling [20]. Our present study demonstrates that SPARC expression upregulated miR-146a as determined by quantitative real time PCR analysis suggesting a possible role of miR-146a in SPARC induced neuronal differentiation.
miR-181 expression was also enhanced in SPARC expressed medulloblastoma cells compared to controls. Several studies have showed that mir-181a and mir-181b act as tumor suppressor genes. mir-181a/b was down-regulated in gliomas and re-introduction of these miRNAs on glioma cell lines results in cell growth inhibition, suppression of invasiveness and induction of apoptosis [60]. miR-181 expression was shown to enhance radio and chemo sensitivity [61-63]. We have previously shown that SPARC enhances radio response in medulloblastoma [22]. Taken together, these findings support the hypothesis that mir-181 may be involved in enhancing radio response in medulloblastoma.
We also demonstrate increased expression of miR-204 in SPARC overexpressed medulloblastoma cells. Several studies have also shown that miR-204-5p is frequently downregulated in papillary thyroid carcinoma, gastric cancer, colorectal cancer, neuroblastoma and endometrial carcinoma, suggesting a common role of miR-204-5p in human tumorigenesis [64-68]. Downregulation of miR-204-5p in human gliomas is correlated with poor patient prognosis, and overexpression of miR-204-5p inhibits the proliferation, migration and invasiveness of glioma cells in vitro and in vivo by directly targeting RAB22A (a member of the RAS oncogene family) [67]. Further, miR-204 was shown to regulate autophagy in renal clear cell carcinoma (RCC) by regulating the expression of LC3 [69]. Our previous studies demonstrated that SPARC expression induced autophagy by modulating LC3 [20, 21]. However, it remains to be determined whether mir-204 induces autophagy in SPARC mediated autophagy in medulloblastoma cells.
Our studies also show that miR-219 is upregulated with SPARC expression. Previous studies demonstrate that miR-219 is downregulated in medulloblastoma tumors and re-introduction of miR-219 inhibits proliferation and suppresses the invasiveness of D283-medulloblastoma cells [70]. Previous data, in combination with our current findings, raise the possibility that induction of miR-219-5p expression may contribute to SPARC mediated suppression of cell proliferation, migration and invasion [20, 22, 23].
Our study also observed that another miR-509-3p associated with cell proliferation and migration is upregulated in SPARC overexpressed cells. miR-509-3p, a significant regulator of the MAP3K8 oncogene was shown to be down regulated in tumor cells [71]. Furthermore, downregulation of miR-509-3p was shown to be a tumor suppressor in tumor cells and associated with tumor cell invasion and migration [72].
We also analyzed the experimentally validated targets of these six miRNAs. Among the validated targets we found transcription factors, cytokines, differentiation, oncogenes, genes involved in histone deacetylation and in cancer cell invasion, apoptosis and angiogenesis. Concurrent with the divergent roles of SPARC in carcinogenesis [15], our study also observed that SPARC altered the expression levels of microRNAs that regulate several signaling pathways involved in tumor progression including toll receptor signaling pathway, interleukin signaling pathway, apoptosis signaling pathway, angiogenesis signaling pathway, inflammation mediated chemokine and cytokine signaling pathway and platelet derived growth factor (PDGF) signaling pathway. Taken together, this established a clear mechanistic interaction underlying miRNA deregulation and tumorigenesis in medulloblastoma cells. Further investigation including functional and mechanistic studies must be undertaken in order to address the biological implications of these miRNAs and to elucidate their involvement in SPARC mediated effects on tumor suppression, treatment response and resistance.
In conclusion, we established that SPARC expression modulated microRNA profile in medulloblastoma cells. Several of the identified miRNAs are involved in tumor progression, suggesting that their deregulated expression may be involved in medulloblastoma tumorigenesis. Studying the targets of deregulated miRNAs genes may elucidate biological functions involved in medulloblastoma pathogenesis.
MATERIALS AND METHODS
Cells and culture condition
Human medulloblastoma cell line D283 was purchased from American Type Culture Collection (ATCC) for this study. D283 cells were authenticated by DNA profile, using the short tandem repeat (STR), cytogenetic analysis, and isoenzymes obtained from ATCC. To obtain consistent results, cells were frozen at third or fourth passage. D283 medulloblastoma cells were cultured in Improved MEM (Zn tonic option medium without phenol red) supplemented with 10 % FBS, 100 units/ml penicillin and 100 µg/ml streptomycin (Gibco BRL, Life Technologies, NY) and collected for use as indicated. Medulloblastoma cells were maintained in a humidified incubator containing 5 % CO2 at 37O C.
Construction of pSPARC and cell transfection
An 1100 bp cDNA human SPARC was amplified by reverse transcription PCR using synthetic primers and cloned into a pcDNA3.1 vector (Invitrogen, San Diego, CA) in sense orientation as described previously [20-23]. Cells were transfected with pcDNA3.1 plasmid containing full length cDNA of SPARC (pSPARC) or empty vector (pEV, a pcDNA vector carrying no vector) using FuGene® HD (Roche, Indianapolis, IN) as per manufacturer’s instructions. For the transfections, cells were cultured in culture flasks (D283) until cells were reached 60-80 % confluence. 7 µg of plasmid DNA were mixed with FuGene® HD in serum free medium per manufacturer’s instructions, and 30 minutes later added to the cells. After allowing 8 hour (h) for optimal transfection, the serum free medium was replaced by complete, supplemented medium, and cells were incubated for 36 h. Cells were harvested for the collection of total RNA and whole cell lysate (total protein) for miRNA profiling with qRT-PCR validation and Western blotting analysis respectively.
Western Blotting Analysis
Medulloblastoma (D283) cells were transfected with mock, pEV or pSPARC for 8 h, and then cells were cells were collected at 36 h post-transfection and lysed in RIPA lysis buffer (50 mM/ml Tris-HCL pH 8.0, 150 mM/ml NaCl, 1 % sodium deoxycholate, 0.1 % SDS) containing 1 mM sodium orthovanadate, 0.5 mM PMSF, 10 mg/ml leupeptin. The protein concentration was determined by Bradford assay [73]. Equal amounts of protein were resolved on SDS-PAGE and then subject to western transfer onto nitrocellulose membranes and kept overnight. The blots were blocked with 5 % nonfat dry milk in TBST (Tris buffer saline with 0.1 % Tween-20) and probed overnight with goat anti-rabbit SPARC polyclonal primary antibody (1:1000; Santa Cruz, CA, USA) and mouse monoclonal p53 primary antibody (1:500; Santa Cruz, CA, USA), followed by incubation with species-specific HRP-conjugated secondary antibodies. An ECL system was used to detect chemiluminescent signals. GAPDH antibody (1:10000; Santa Cruz, CA, USA) was used to verify equal amounts of proteins were loaded in all lanes.
MicroRNA (miRNA) microarray and analysis
Total RNA was extracted from D283 cells using TRIZOL reagent (Invitrogen, Carlsbad, CA) as per standard control protocol. Quality control of the total RNA samples was assessed using UV Spectrophotometry and agarose gel electrophoresis. The samples were DNase digested and low-molecular weight (LMW) RNA was isolated by ultrafiltration through YM-100 columns (Millipore) and subsequent purification using the RNeasy MinElute Clean-Up Kit (Qiagen Inc., Valencia, CA). RNA samples (Control (pEV control), SPARC-overexpression and SPARC-underexpression D283 medulloblastoma cells) were processed at Ocean Ridge Bioscience (ORB, Palm Beach Gardens, FL, USA) for analysis using custom multi-species microarrays containing 1209 human mature probes miRNAs present in the miRBase version 16.0 database. An empty vehicle (pEV) control and SPARC overexpressed or SPARC underexpressed samples were assessed in biological triplicate. The sensitivity of microarray is such that it could detect as low as 20 amoles of synthetic miRNA being hybridized along with each sample. The microarrays were produced by Microarrays Inc. (Huntsville, Alabama), and made up of epoxide glass substrates that had been spotted in triplicate with each probe.
miRNA sample processing
The LMW RNA samples were 3’-end labelled with Oyster-550 fluorescent dye using the Flash Taq RNA labelling Kit (Genisphere, Hatfield, PA). Labelled RNA samples were then hybridized to the miRNA microarrays according to the conditions outlined in the Flash Taq RNA labeling Kit protocol. The microarrays were scanned on an Axon Genepix 4000B scanner, and data was extracted from image using GenePix V4.1 software.
Data pre-processing
Spot intensities were obtained for the 8816 features on each microarray by subtracting the medial local background from the median local foreground for each spot. The spot intensities and 95th percentile of negative control spot was also calculated for each array. The spot intensities and 95th percentile of negative controls (TPT95) were transformed by taking the Logarithm base 2 (indicated as Log2) of each value. The normalization factor (N) for each microarray was determined by achieving the 20 % trimmed mean of the human probe intensities that were detected one Log2 unit above TPT95 (TPT95 + 1) in all samples and with standard deviation of probe intensities among all samples less than 1.25. The Log2 transformed spot intensities for all 8816 features were normalized, by subtracting N from each spot intensity and scaled by adding the grand mean of N across all microarrays. The mean probe intensities for each of the 1209 human probes on each of the 20 arrays were then determined by averaging the triplicate spot intensities. Spots flagged as poor quality during data extraction were omitted prior to averaging. The 1209 human non-control Log2 transformed, normalized and averaged probe intensities were filtered to obtain a panel of 729 human miRNA probes showing probe intensity greater than one log2 unit above TPT95 (> TPT95 + 1) in at least 10 % of the samples.
Quality control
Sensitivity of the microarray hybridization was confirmed by detection of hybridization signal for all 11 spikes that were added during isolation above the detection threshold level (labelling well above TPT95). The array also contains a set of specificity control probes complementary to three different miRNAs. Each specificity control includes a perfect match, signal mismatch, double mismatch and shuffled version of probes. Reproducibility of the arrays was determined by monitoring the hybridization intensity for the triplicate human spots on each array. Differential expression analysis
For statistical analysis, samples were binned in two groups: control and SPARC-overexpression or SPARC-underexpression. The Log2 transformed and normalized spot intensities for the 623 detectable were examined for difference between the groups by at least 2-fold difference between the control and SPARC-overexpression or SPARC-underexpression. A total 27 differentially expressed probes exhibited at-least two-fold difference between the control and SPARC-overexpression and/or SPARC-underexpression samples.
Hierarchical clustering of miRNA array data
Data for the total 729 detectable human probes were clustered using Cluster 3.0 Software [74]. Genes were median-centered prior to hierarchical clustering. Hierarchical clustering was conducted using Centered Correlation as the similarity metric and Average Linkage as the clustering method. Intensity scale is shown in arbitrary units.
Prediction of miRNA targets
To predict mRNA targets, a total 27 differentially expressed miRNAs which were identified in our analysis by looking at the at least 2-fold change difference between the control and SPARC-overexpression or SPARC-underexpression samples. These 27 miRNAs were uploaded into the Ingenuity Pathway Analysis (IPA) web-based tool (Ingenuity Systems, Redwood City, CA). Putative miRNA-mRNA relationships were identified using the IPA microRNA Target Filter, based on a knowledgebase of predicted and experimentally observed relationships. This database included interactions captured by Ingenuity Systems curators, as well as interactions from two external databases namely TarBase and miRecords. Furthermore, TarBase is a database that represents a comprehensive collection of miRNA targets with experimental support from published research articles. The interaction in TarBase are curated from both disease and non-disease related studies that use method to increase or decrease the expression of a particular miRNA of interest and evaluate its downstream transcriptional effects [75]. miRecords is a database of both experimentally validated miRNA targets that were captured through the compilation of findings from 2,705 studies, to date [76]. Moreover, the majority of the information in miRecords is collected from low-throughput experiments that also involve altering the expression levels of particular miRNAs in animal based experimental studies [76]. We stringently selected for only the experimentally observed miRNA-mRNA relationships, and the resulting target gene list was analyzed for functional network and biological pathway analysis using the PANTHER web tool (http://www.pantherdb.org/) [77]. The list of target genes was compared to a reference list to statistically determine over- or under-representation of PANTHER classification categories. p-values were calculated with Bonferroni correction test and p < 0.05 was considered to estimate if a specific PANTHER category was over-or under- represented in a statistically significant way.
Network analysis
The biologically network analyses were performed using IPA software. These mRNA targets with their respective miRNAs overlaid onto a global interaction network. Networks containing miRNA signaling where algorithmically evaluated by a modified Fisher’s exact test [78]. Biological functions and disease signatures within the constructed networks were then identified. Overrepresented functions/diseases were defined as those that contain more mRNA targets than expected by chance, as calculated by the right-tailed Fisher’s exact test with a p value < 0.05 considered as a statistically significant.
Data mining using the Oncomine Database for six miRNAs predicted gene targets
The expression pattern of six miRNAs predicted gene targets in desmoplastic medulloblastoma versus normal control samples were identified from Oncomine Database (www.oncomine.org/ Compendia Bioscience, Ann Arbor, MI, USA) [39, 40]. Oncomine is an online database that contains manually curated microarray and gene copy number expression data from various studies related to cancer. Currently, Oncomine Database contains data from more than 18,000 cancer gene expression microarray data from over 500 cancer types including human tumor samples and human cancer cell lines [39, 40]. The Oncomine platform allows us to compare gene expression across multiple cancer studies to identify genes that are differentially over or under expressed in different cancer types. Herein, we mined the microarray-gene expression data of human desmoplastic medulloblastoma cancer type samples versus normal control samples to identify six miRNAs predicted gene targets of SPARC overexpressed medulloblastoma cell samples versus control cell samples.
miRNA qRT-PCR validation of six differentially expressed miRNAs in medulloblastoma
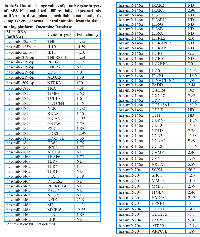
We tested the expression of levels of six miRNAs (has-miR-125b-1*, has-miR-146a-5p, has-miR-181a-5p, has-miR-204-5p, has-miR-219-5p and has-miR-509-3p) which were found to be differentially regulated with SPARC expression in medulloblastoma cells. Total RNA was extracted from D283 cells transfected with pSPARC or pEV using a miRNeasy Mini Plus kit (Qiagen, Valencia, CA) with QIAzol Lysis Reagent (Qiagen, Valencia, CA) for cell lysis. The RNA isolation procedure was accomplished using manufacture’s instruction. 1 µg of total RNA was used as a template for reverse transcription reaction using the miScript II Reverse Transcription kit (Qiagen, Valencia, CA), accordingly to the manufacturer’s instructions using 5X miScript HiFlex Buffer, which enables conversion of mature as well as precursor miRNAs. qRT-PCR was performed in 25 µl reaction using the miScript SYBR Green PCR kit (Qiagen, Valencia, CA), miRNA forward primers (primer sequences are presented in Table 1), and the reference U6 housekeeping miRNA in combination with the miScript Universal reverse primer (supplied with miScript SYBR Green PCR kit) for the cDNA templates. qRT-PCR reactions were performed in triplicate and independently repeated at least twice with no template control on 7500HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). PCRs were processed through an initial denaturation at 95O C for 15 min and by 40 cycles of 3-step PCR, including 15 sec of denaturation at 94O C, a 30 sec annealing phase at 55O C and an elongation phase at 70O C for 34 sec. The resulting RT-PCR cycle times were normalized against the U6 housekeeping miRNA, and fold change (FC) values of miRNA expression was calculated using the ∆∆Ct method. Fold changes compared to controls were calculated by 2-∆∆Ct after normalizing to U6, a reference gene. Statistical significance of the difference in miRNA expression levels between the empty vehicle (pEV) control and SPARC-overexpression (pSPARC) was calculated by unpaired t-tests in Graphpad Prism 5 Software (Graphpad Software Inc., San Diego, CA).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from D283 mock or pEV control and pSPARC cells using TRIzol ® solution (Invitrogen, Carlsbad, CA) as per standard protocol. For cDNA synthesis total RNA (1 µg) of was reverse transcribed with high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA), and subsequently used to amplify SPARC, p53 and p21 using iTaqTM Universal SYBR® Green Supermix kit (Bio-Rad Laboratories, Hercules, CA). GAPDH was used as a housekeeping reference gene in this analysis. qRT-PCR reaction mix was performed using the CFX96TM Real-Time PCR System (Bio-Rad Laboratories, Hercules, CA) with following primers: SPARC forward, 5’-ATC TAA ATC CAC TCC TTC CAC AG-3’ and reverse, 5’-CAC CGT TAA TGT ATT CAC TTA AAT C-3’; p53 forward, 5’-ACA CGC TTC CCT GGA TTG-3’ and reverse, 5’-TCG ACG CTA GGA TCT GAC TG-3’; p21 forward, 5’-GAG GCC GGG ATG AGT TGG GAG GAG-3’ and reverse, 5’-CAG CCG GCG TTT GGA GTG GTA GAA-3’ and GAPDH forward, 5’-GAA GGT GAA GGT CGG AGT C-3’ and reverse, 5’-GAA GAT GGT GAT GGG ATT TC-3’. Fold changes between the pSPARC and the control group (mock or pEV) were calculated using delta-delta cycle threshold (ΔΔCt) values and normalized with GAPDH as a housekeeping gene. Statistical significance of the transcript levels between pSPARC versus the control group was calculated using an unpaired t-test in Graphpad Prism 5 Software (Graphpad Software Inc., San Diego, CA).
ACKNOWLEDGEMENTS AND FUNDING
This work was supported by the University of Illinois Cancer Center, Section of Hematology and Oncology, Department of Medicine, Chicago, IL 60612, USA.
CONFLICT OF INTEREST
All authors do not have any conflict of interest, do not own, receive an income or have been financed from a private or public company.

- 1. Crawford JR, MacDonald TJ and Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007; 6:1073-1085. [PubMed]
- 2. Radiation induced adult medulloblastoma: a case report. J Neurooncol. 2006; 80:191-194. [PubMed]
- 3. Current treatment of medulloblastoma: recent advances and future challenges. Semin Oncol. 2004; 31:666-675. [PubMed]
- 4. Results of a prospective randomized trial comparing standard dose neuraxis irradiation (3,600 cGy/20) with reduced neuraxis irradiation (2,340 cGy/13) in patients with low-stage medulloblastoma. A Combined Children’s Cancer GroupPediatric Oncology Group Study. Pediatr Neurosurg. 1996; 24:167-176. [PubMed]
- 5. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97-109. [PubMed] https://doi.org/10.1007/s00401-007-0243-4.
- 6. The molecular genetics of medulloblastoma: an assessment of new therapeutic targets. Neurosurg Rev. 2008; 31:359-368. [PubMed]
- 7. Medulloblastoma: is the 5-year survival rate improving? A review of 80 cases from a single institution. J Neurosurg. 1997; 86:13-21. [PubMed]
- 8. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012; 482:529-533. [PubMed] https://doi.org/10.1038/nature10825.
- 9. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002; 14:608-616. [PubMed]
- 10. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001; 19:816-827. [PubMed]
- 11. Structure of a novel extracellular Ca(2+)binding module in BM-40. Nat Struct Biol. 1996; 3:67-73. [PubMed]
- 12. The biology of SPARC, a protein that modulates cell-matrix interactions. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1994; 8:163-173. [PubMed]
- 13. 2 regulation of extracellular matrix and cell cycle in mice lacking the matricellular protein SPARC. You can believe your eyes. J Histochem Cytochem. 1999; 47:1643A-1643. [PubMed]
- 14. Diverse biological functions of the SPARC family of proteins. Int J Biochem Cell Biol. 2012; 44:480488. [PubMed] https://doi.org/10.1016/j.biocel.2011.12.021.
- 15. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis. 2014; 35:967-973. [PubMed]
- 16. Modulation of SPARC expression during butyrate-induced terminal differentiation of cultured human keratinocytes: regulation via a TGF-beta-dependent pathway. Exp Cell Res. 1993; 206:261-275. [PubMed]
- 17. Recombinant mouse SPARC promotes parietal endoderm differentiation and cardiomyogenesis in embryoid bodies. Biochem Cell Biol. 2008; 86:487-499. [PubMed]
- 18. Involvement of SPARC in in vitro differentiation of skeletal myoblasts. Biochemical and biophysical research communications. 2000; 271:630-634. [PubMed]
- 19. Haemocyte-derived SPARC is required for collagen-IVdependent stability of basal laminae in Drosophila embryos. J Cell Sci. 2008; 121:1671-1680. [PubMed]
- 20. SPARC stimulates neuronal differentiation of medulloblastoma cells via the Notch1/STAT3 pathway. Cancer research. 2011; 71:4908-4919. [PubMed] https://doi.org/10.1158/0008-5472.CAN-10-3395.
- 21. Cathepsin B facilitates autophagymediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010; 17:1529-1539. [PubMed] https://doi.org/10.1038/cdd.2010.28.
- 22. Blockade of SOX4 mediated DNA repair by SPARC enhances radioresponse in medulloblastoma. Cancer letters. 2012; 323:188-198. [PubMed] https://doi.org/10.1016/j.canlet.2012.04.014.
- 23. SPARC expression induces cell cycle arrest via STAT3 signaling pathway in medulloblastoma cells. Biochemical and biophysical research communications. 2012; 417:874879. [PubMed] https://doi.org/10.1016/j.bbrc.2011.12.065.
- 24. Signaling pathways in medulloblastoma. J Cell Physiol. 2008; 217:577-583. [PubMed]
- 25. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008; 27:523-537. [PubMed]
- 26. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297. [PubMed]
- 27. Identification of active transcription factor and miRNA regulatory pathways in Alzheimer’s disease. Bioinformatics. 2013; 29:25962602. [PubMed]
- 28. Regulation of Myelination in the Central Nervous System by Nuclear Lamin B1 and Non-coding RNAs. Transl Neurodegener. 2014; 3:1-4. [PubMed] https://doi.org/10.1186/2047-9158-3-4.
- 29. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92-105. [PubMed] https://doi.org/10.1101/gr.082701.108.
- 30. Epigenomics of Alzheimer’s disease. Transl Res. 2015; 165:200-220. [PubMed] https://doi.org/10.1016/j.trsl.2014.05.006.
- 31. Systems approach to neurodegenerative disease biomarker discovery. Annu Rev Pharmacol Toxicol. 2014; 54:457-481. [PubMed]
- 32. MicroRNAs: molecular features and role in cancer. Front Biosci (Landmark Ed). 2012; 17:2508-2540. [PubMed]
- 33. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006; 6:259-269. [PubMed]
- 34. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:2257-2261. [PubMed] https://doi.org/10.1073/pnas.0510565103.
- 35. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer research. 2005; 65:6029-6033. [PubMed]
- 36. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochemical and biophysical research communications. 2005; 334:1351-1358. [PubMed]
- 37. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005; 353:1793-1801. [PubMed]
- 38. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39(Database issue):D152-157. [PubMed] https://doi.org/10.1093/nar/gkq1027.
- 39. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007; 9:166-180. [PubMed]
- 40. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004; 6:1-6. [PubMed]
- 41. miR-let-7f-1 regulates SPARC mediated cisplatin resistance in medulloblastoma cells. Cellular signalling 2014; 26:2193-2201.. 2004; 6:1-6. [PubMed]
- 42. A differentially expressed set of microRNAs in cerebro-spinal fluid (CSF) can diagnose CNS malignancies. Oncotarget. 2015; 6:20829-20839. [PubMed] https://doi.org/10.18632/oncotarget.4096.
- 43. MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol Cell Biol. 2009; 29:5290-5305. [PubMed] https://doi.org/10.1128/MCB.01694-08.
- 44. miR-125b inhibits Connexin43 and promotes glioma growth. Cellular and molecular neurobiology. 2013; 33:1143-1148. [PubMed]
- 45. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009; 23:862-876. [PubMed] https://doi.org/10.1101/gad.1767609.
- 46. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2009; 23:347-358. [PubMed]
- 47. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 2008; 27:2616-2627. [PubMed] https://doi.org/10.1038/emboj.2008.172.
- 48. MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain research. 2010; 1312:120-126. [PubMed]
- 49. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012; 26:2639-2647. [PubMed]
- 50. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR125b. The Journal of biological chemistry. 2007; 282:14791486. [PubMed]
- 51. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer research. 2011; 71:3552-3562. [PubMed]
- 52. Micro-RNA regulation of the mammalian lin-28 gene during neuronal differentiation of embryonal carcinoma cells. Mol Cell Biol. 2005; 25:91989208. [PubMed] https://doi.org/10.1128/MCB.25.21.9198-9208.2005.
- 53. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the downregulation of putative targets during differentiation. The Journal of biological chemistry. 2005; 280:16635-16641. [PubMed]
- 54. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol. 2011; 31:3584-3592. [PubMed] https://doi.org/10.1128/MCB.05821-11.
- 55. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008; 27:5643-5647. [PubMed] https://doi.org/10.1038/onc.2008.171.
- 56. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer research. 2009; 69:1279-1283. [PubMed] https://doi.org/10.1158/0008-5472.CAN-08-3559.
- 57. miR-146a suppresses invasion of pancreatic cancer cells. Cancer research. 2010; 70:14861495. [PubMed] https://doi.org/10.1158/0008-5472.CAN-09-2792.
- 58. Loss of mir-146a function in hormone-refractory prostate cancer. RNA. 2008; 14:417-424. [PubMed] https://doi.org/10.1261/rna.874808.
- 59. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010; 142:914-929. [PubMed] https://doi.org/10.1016/j.cell.2010.08.012.
- 60. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain research. 2008; 1236:185-193. [PubMed]
- 61. MiR-181b suppresses proliferation of and reduces chemoresistance to temozolomide in U87 glioma stem cells. J Biomed Res. 2010; 24:436-443. [PubMed] https://doi.org/10.1016/S1674-8301(10)60058-9.
- 62. Non-coding MicroRNAs hsa-let7g and hsa-miR-181b are Associated with Chemoresponse to S-1 in Colon Cancer. Cancer Genomics Proteomics. 2006; 3:317-324. [PubMed]
- 63. MiR-181b sensitizes glioma cells to teniposide by targeting MDM2. BMC Cancer. 2014; 14:611. [PubMed] https://doi.org/10.1186/1471-2407-14-611.
- 64. A TrkB-STAT3-miR204-5p regulatory circuitry controls proliferation and invasion of endometrial carcinoma cells. Mol Cancer. 2013; 12:155. [PubMed] https://doi.org/10.1186/1476-4598-12-155.
- 65. MiR-204-5p suppresses cell proliferation by inhibiting IGFBP5 in papillary thyroid carcinoma. Biochemical and biophysical research communications. 2015; 457:621-626. [PubMed]
- 66. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br J Cancer. 2012; 107:967976. [PubMed] https://doi.org/10.1038/bjc.2012.356.
- 67. miR204-5p inhibits proliferation and invasion and enhances chemotherapeutic sensitivity of colorectal cancer cells by downregulating RAB22A. Clin Cancer Res. 2014; 20:61876199. [PubMed]
- 68. MicroRNA-204-5p inhibits gastric cancer cell proliferation by downregulating USP47 and RAB22A. Med Oncol. 2015; 32:331. [PubMed]
- 69. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012; 21:532-546. [PubMed] https://doi.org/10.1016/j.ccr.2012.02.019.
- 70. miR-219 inhibits the proliferation, migration and invasion of medulloblastoma cells by targeting CD164. Int J Mol Med. 2014; 34:237-243. [PubMed]
- 71. MicroRNA-509-3p inhibits cancer cell proliferation and migration by targeting the mitogen-activated protein kinase kinase kinase 8 oncogene in renal cell carcinoma. Mol Med Rep. 2015; 12:1535-1543. [PubMed]
- 72. Identification of miR-508-3p and miR-5093p that are associated with cell invasion and migration and involved in the apoptosis of renal cell carcinoma. Biochemical and biophysical research communications. 2012; 419:621-626. [PubMed]
- 73. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72:248-254. [PubMed]
- 74. Open source clustering software. Bioinformatics. 2004; 20:1453-1454. [PubMed]
- 75. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 2009; 37(Database issue):D155-158. [PubMed] https://doi.org/10.1093/nar/gkn809.
- 76. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009; 37(Database issue):D105-110. [PubMed] https://doi.org/10.1093/nar/gkn851.
- 77. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013; 41(Database issue):D377-386. [PubMed] https://doi.org/10.1093/nar/gks1118.
- 78. A network-based analysis of systemic inflammation in humans. Nature. 2005; 437:1032-1037. [PubMed]
- 79. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. The Journal of biological chemistry. 2012; 287:2608-2617. [PubMed] https://doi.org/10.1074/jbc.M111.285718.
- 80. miR-125b, a target of CDX2, regulates cell differentiation through repression of the core binding factor in hematopoietic malignancies. The Journal of biological chemistry. 2011; 286:3825338263. [PubMed] https://doi.org/10.1074/jbc.M111.269670.
- 81. microRNA-125b inhibits cell migration and invasion by targeting matrix metallopeptidase 13 in bladder cancer. Oncology letters. 2013; 5:829-834. [PubMed] https://doi.org/10.3892/ol.2013.1123.
- 82. MUCIN 1 ONCOPROTEIN EXPRESSION IS SUPPRESSED BY THE miR-125b ONCOMIR. Genes & cancer. 2010, 1:62-68. [PubMed] https://doi.org/10.1177/1947601909357933.
- 83. MicroRNA-125b down-regulation mediates endometrial cancer invasion by targeting ERBB2. Medical science monitor : international medical journal of experimental and clinical research. 2012; 18:BR149-155. [PubMed] https://doi.org/10.12659/MSM.882617.
- 84. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene. 2013, 32:3071-3079. [PubMed]
- 85. Regulation of placenta growth factor by microRNA-125b in hepatocellular cancer. Journal of hepatology. 2011; 55:1339-1345. [PubMed] https://doi.org/10.1016/j.jhep.2011.04.015.
- 86. miR-125b suppresses the proliferation and migration of osteosarcoma cells through down-regulation of STAT3. Biochemical and biophysical research communications. 2011; 416:31-38. [PubMed]
- 87. Roles and mechanism of miR-199a and miR125b in tumor angiogenesis. PloS one. 2013; 8:e56647. [PubMed] https://doi.org/10.1371/journal.pone.0056647.
- 88. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene. 2013, 32:2984-2991. [PubMed]
- 89. Oncomir miR-125b suppresses p14(ARF) to modulate p53-dependent and p53-independent apoptosis in prostate cancer. PloS one. 2013; 8:e61064. [PubMed] https://doi.org/10.1371/journal.pone.0061064.
- 90. The role of microRNA genes in papillary thyroid carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:19075-19080. [PubMed] https://doi.org/10.1073/pnas.0509603102.
- 91. Aberrant expression of oncogenic and tumorsuppressive microRNAs in cervical cancer is required for cancer cell growth. PloS one. 2008; 3:e2557. [PubMed] https://doi.org/10.1371/journal.pone.0002557.
- 92. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010; 115:265-273. [PubMed]
- 93. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PloS one. 2013; 8:e60317. [PubMed] https://doi.org/10.1371/journal.pone.0060317.
- 94. miR-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. The Prostate. 2014; 74:1613-1621. [PubMed]
- 95. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer letters. 2016; 389:11-22. [PubMed]
- 96. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer research. 2015; 75:26742685. [PubMed] https://doi.org/10.1158/0008-5472.CAN-14-2875.
- 97. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma. 2010; 57:264-269. [PubMed]
- 98. LncRNAUCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Scientific reports. 2016; 6:23892. [PubMed] https://doi.org/10.1038/srep23892.
- 99. miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer. Cell death & disease. 2012; 3:e423. [PubMed] https://doi.org/10.1038/cddis.2012.160.
- 100. MiR-219-5p inhibits hepatocellular carcinoma cell proliferation by targeting glypican-3. FEBS letters. 2012; 586:884-891. [PubMed]
- 101. miR-219-5p modulates cell growth of papillary thyroid carcinoma by targeting estrogen receptor alpha. The Journal of clinical endocrinology and metabolism. 2015; 100:E204213. [PubMed] https://doi.org/10.1210/jc.2014-2883.
- 102. miR-5093p is clinically significant and strongly attenuates cellular migration and multi-cellular spheroids in ovarian cancer. Oncotarget. 2016; 7:25930-25948. [PubMed] https://doi.org/10.18632/oncotarget.8412.
Last Modified: 2017-03-06 21:34:09 EST
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.