Genes & Cancer
Abstract | PDF | Full Text | Supplementary Materials | Press Release
https://doi.org/10.18632/genesandcancer.239
Analysis of pathogenic variants in retinoblastoma reveals a potential gain of function mutation
Ana María Peña-Balderas1, Mayra Martínez-Sánchez1, Isaí Olmos-Sánchez1, Karla Calderón-González1, Mariana Moctezuma-Dávila1,4, Martha Rangel-Charqueño2, Jesús Hernández-Monge3 and Vanesa Olivares-Illana1
1Laboratorio de Interacciones Biomoleculares y Cáncer, Instituto de Física Universidad Autónoma de San Luis Potosí, San Luis Potosí 78210, México
2División de Cirugía, Departamento de Oftalmología, Hospital Central “Ignacio Morones Prieto”, San Luis Potosí, México
3Investigador por México, Laboratorio de Biomarcadores Moleculares, Instituto de Física, Universidad Autónoma de San Luis Potosí, México City, México
4Present address: Houston Methodist Hospital, Department of Pathology and Genomic Medicine, Houston, TX 77030, USA
Correspondence to: Vanesa Olivares-Illana and Jesús Hernández-Monge, email: vanesa@ifisica.uaslp.mx, jesus_hernandez_m@hotmail.com
Keywords: retinoblastoma; cancer; gain of function; mutants; pathogenic variants
Received: October 05, 2024
Accepted: January 09, 2025
Published: January 20, 2025
Copyright: © 2025 Peña-Balderas et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ABSTRACT
Retinoblastoma (Rb1) is a gene that codes for a tumour suppressor protein involved in various types of cancer. It was first described in retinoblastoma and is segregated as an autosomal dominant trait with high penetrance. In 1971, Knudson proposed his hypothesis of the two hits, where two mutational events are required to initiate tumour progression. We analysed three different point mutations present in patients’ retinoblastoma. We produced three cell lines with retinoblastoma protein (RB) mutated in various regions: the missense pN328H, pD718N, and the nonsense early stop codon pR552*. We studied the effect of these point mutations on levels of mRNA and protein expression, proliferation, viability, localisation, and migration using an RBKO cell line. All three affected their localisation patterns and proliferation. However, the pR552* mutation also increases viability and migration. Moreover, when this mutation is simultaneously expressed with a wild-type RB, the phenotype and proliferation parameters are as with the mutant alone, suggesting that maybe only one mutated allele is needed to trigger the characteristic cancer phenotype. In other words, the pR552* mutant behaves more like a gain-of-function or oncogenic mutant. Indeed, a family carrying this mutation showed complete penetrance and high expressivity.
INTRODUCTION
Retinoblastoma, also known as retina cancer, is a rare pediatric cancer that occurs among children aged 0 to 5 years old. It is the most frequent ocular cancer, accounting for 4% of all pediatric neoplasms [1]. Nevertheless, the incidence varies depending on the region; for example, in non-developed countries, it can increase up to 24 cases per million of habitants [2, 3]. Retinal tumours’ are commonly detected by visually noticeable signs such as leukocoria (white pupil) or strabismus (misaligned eyes), different-coloured irises, and poor vision, among others [4]. Retinoblastoma is curable as long as it is detected and treated early; if not, the tumour tends to invade and destroy internal structures of the eye globe, eventually leading to the development of metastasis and death [5]. The Rb1 gene was the first tumour suppressor identified and cloned; its association with the development of retinal cancer gave rise to its name, “RB”, for retinoblastoma [6]. Although the loss of Rb1 plays a central role in the development of retinoblastoma, other genes are also protagonists, such as Myc-N, Mdm2, Mdmx, p53, and others recently identified [5, 7].
Many authors have annotated several alterations in the Rb1 gene, not only for retinoblastoma but also for other cancers [8–11]. The mutations reported on the Rb1 gene are widely distributed across all 27 exons and their regulatory regions [12]. Missense variants, synonymous mutants, early stop codons (nonsense), and deletions, among others, have been reported [13]. More than one hundred different mutations have been identified and reported in the Rb1 gene associated with retinoblastoma in the COSMIC database: https://cancer.sanger.ac.uk/cosmic [14]. It has been suggested that unlike other types of cancers, retinoblastoma tumours do not exhibit large chromosomal rearrangements but rather punctual changes scattered throughout the gene [4]. Not all mutations cause the same phenotype; for instance, it has been observed that early stop codons are associated with high penetrance of the disease; missense mutations, small in-frame indels, promoter and some splicing mutations are associated with low penetrance phenotype [11]. The RB protein, encoded by the Rb1 gene, is a large polypeptide consisting of 928 amino acid residues that undergo post-translational modifications. It comprises four domains with intrinsically disordered regions, providing the protein with significant plasticity. These characteristics allow RB to have a broad range of interaction partners [15, 16]. This network of interactions, where RB functions as a hub, allows RB to be involved in many different pathways. A mutation in Rb1 could interfere with various interactions and post-translational modifications, among other processes [6].
It is widely accepted that the factor that triggers this type of cancer is the homozygous loss of the Rb1 gene, according to Knudson’s two-hits hypothesis [17, 18]. However, with the tumour suppressor p53 exists strong evidence showing that a cancer phenotype can be triggered with one single mutation of p53 in one allele. Bernard et al. showed that from 3324 patients with myelodysplastic syndrome, one-third presented monoallelic mutations of p53 [19]. This is in line with other reports where heterozygous inactivation of p53 in rats develop sarcomas at eight months of age [20]. In addition, there is recent evidence in retinoblastoma tumours itself where only one allele was found mutated [21, 22].
Despite these abundant reports, none have further explored the underlying mechanisms by which RB mutants perturb cellular homeostasis. In this work, we aimed to explore the impact of a nonsense pR552* mutation and missense pN328H and pD718N mutations in the Rb1 gene on cellular activities such as cell proliferation, migration or subcellular localization. We observed a mislocalization of all three mutants. Furthermore, the pR552* mutant showed the highest growth rate, viability, and migration compared to its wild-type counterpart, either alone or co-expressed with the wild-type. This finding raises the question of whether, similar to the tumour suppressor protein p53, it is also possible to find gain-of-function mutants in RB.
RESULTS
RB mutants found in retinoblastoma patients
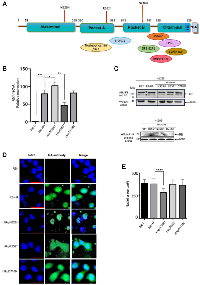
Retinoblastoma (RB) is a phosphoprotein consisting of 928 amino acid residues and comprising various domains. It begins with the N-terminal domain, followed by pocket A and pocket B, and concludes with the C-terminal domain (Figure 1A). The multidomain structure of this protein provides it with significant plasticity, enabling it to interact with a vast array of different partners [16, 23–26] (Figure 1A). Rb1 undergoes mutations in several types of cancers, including retina, lung, prostate cancer, osteosarcoma, glioma, among others [6, 7, 27–30]. Here, through site-directed mutagenesis, we generated three distinct mutations identified in retinal cancer patients using an RB template labeled RB-HA, which incorporates an HA-tag at the 3′-end. One of these mutants, pR552* is also prevalent in other types of cancers (Table 1). The first mutation, pN328H, represents a conservative point mutation. According to alpha fold simulation, it exhibits a slight conformational change in the loop between pockets A and B, and a significant alteration in the C-terminal domain [31] (Supplementary Figure 1A). The second mutant is pD718N, representing a non-conservative mutation. The alpha fold simulation indicates a slight conformational change in the N-terminal loop, the loop between pockets A and B, and a pronounced change in the C-terminal domain (Supplementary Figure 1B). Lastly, we generated a mutant of pR552* that introduces a stop codon at residue R552 located in pocket A. This mutant has been identified not only in retinoblastoma but also in osteosarcoma, glioma, among other types of cancer. The alpha fold simulation suggests a slight conformational change in the N-terminal loop; it is worth noting that this mutant lacks the pocket B and the C-terminal domain (Supplementary Figure 1C). The first two mutants exhibit changes in the C-terminal region; nonetheless, they potentially interact with some of the proteins that recognize the sequence of this domain. In contrast, pR552* completely lacks this region, losing the ability to interact with a group of proteins involved in various signaling pathways, including crucial players such as E2F1 and cyclin A.
Cellular expression and localization of the three different RB mutants
To investigate the phenotype of these mutations, we initially knocked out the endogenous RB protein using CRISPR/Cas in the H1299 cell line, which lacks p53 expression (Supplementary Figure 2). We chose H1299 to eliminate p53-dependent apoptosis unleashed by RB absence. First, we stably transfected the H1299 RB−/− cells with each mutant, determined the Rb1 mRNA expression levels, and compared them with the Rb1-HA mRNA version and the three mutants (Figure 1B). In terms of mRNA expression, we observed that the mutant HA-D718N did not exhibit a change compared to Rb1-HA, the mutant HA-N328H showed increased expression levels, and finally, the mutant HA-R552* showed a significantly decreased expression (Figure 1C). In contrast, at the protein level, the mutant HApN328H show a minimal level of expression, whereas the HApD718N mutant shows a similar level of expression in comparison with the RB-HA. The HApR552* showed an important decrease in protein expression in line with the mRNA levels (Figure 1C).
Immunofluorescence assays were performed on each cell line generated for the mutants. An anti-HA-tag antibody was employed to detect each of the RB proteins. The HApN328H mutant exhibits nuclear localization similar to the wild-type RB-HA. Interestingly, this mutant shows a smaller nucleus size and cannot localize into the nucleoli. It has been shown that hyperphosphorylated RB can interact with nucleophosmin/B23 through the RB pocket A region [32]. According to the alpha fold simulation, the pN328H mutant exhibits a change in the intrinsic disorder region between pocket A and B, potentially explaining its inability to translocate to the nucleolus. Alternatively, the mutation may promote a change in the phosphorylation pattern (Figure 1A, 1D and Supplementary Figure 1A).
On the other hand, mutants HApD718N and HApR552* lose the localization pattern and are found in both the cytoplasm and the nucleus. It has been reported that the nuclear localization signal of RB is located in the C-terminal region; the mutant pD718N shows a significant change in the conformation of this region, whereas pR552* lacks the entire domain. The nucleus sizes remain identical to the wild-type RB-HA (Figure 1A, 1E and Supplementary Figure 1B, 1C).
Cell proliferation, migration and viability
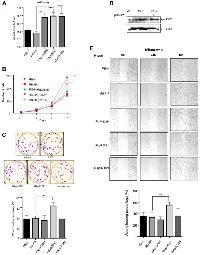
To assess the proliferation rate of the stably transfected cells for each mutant, we conducted the colorimetric MTT assay. We observed that all three mutants exhibited an increase in proliferation. Notably, the mutant HApD718N and the smaller mutant HApR552* showed the highest increase in proliferation according to the MTT assay (Figure 2A). These findings were further confirmed with a cell counting assay; in this experiment, HApR552* exhibited the highest proliferation rate, more than twice that of the wild-type RB-HA (Figure 2B). HApD718N also displayed a high proliferation rate, while HApN328H showed a slight increase in proliferation, barely higher than the wild type (Figure 2B).
In addition, we performed a clonogenic assay, also known as a colony formation assay, which is based on the ability of a single cell to grow into a colony. This assay quantitatively measures the ability to undergo unlimited division and serves as an indicator of cancer cells [33, 34]. As expected, all mutant cell lines generated colonies, as H1299 is indeed a cancer cell line. Interestingly, only the mutant HApR552*, which produces a truncated protein, showed a significant increase in its ability to form colonies (Figure 2C). Notably, this specific mutant lacks the C-terminal region, thereby also losing the ability to interact with proteins as the transcription factor E2F1 and potentially the capacity to arrest the cell cycle in the G1/S phase.
Some reports [35, 36] have shown that in the absence of the wild-type RB protein, its homologous RBL1/p107 increases its levels to compensate for the loss of cell cycle control. We investigated whether, in the presence of the HApR552*, the RBL1/p107 also increased its levels. Indeed, our observations indicated that RBL1/p107 increases in the presence of HApR552* (Figure 2D). It is possible that the expression of the truncated form HApR552* could, in some way, interfere with the role of RBL1/p107 through the binding of common targets, at least via their N-terminal domains.
Finally, a wound-healing assay was employed to evaluate the migration capacity of investigated cell lines. We observed a significant increase in wound healing with the HApR552* mutant. However, none of the other mutants exhibited any effect on the migration assay (Figure 2E). Taken together, these results indicate that the HApR552* exhibits the most significant perturbation in behavior compared to the RB-HA wild-type protein. This mutant pR552* has been identified with high recurrence in different studies in Vietnamese [37], Portuguese [38], Canadian [39], American [13], English [11], and Mexican (this work) patients among others; consequently, we decided to continue studying this particular mutant.
Effect of the HA tag on the behavior of the truncated HApR552* mutant
The truncated mutant HApR552* shows high recurrence in patients; it also exhibits the most significant impact on the cell at various levels, including localization, migration, proliferation, and viability. Consequently, we questioned whether the HA-tag plays a role in the observed effects of the mutant. To address this, we established a stable cell line with the truncated mutant lacking the HA-tag, named pR552*, and assessed its impact on the cell phenotype.
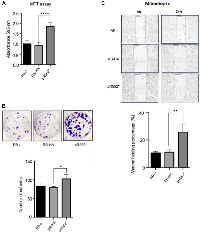
To examine proliferation, we conducted MTT experiments. Notably, the difference in proliferation between HApR552* and pR552* compared to RB-HA is highly pronounced and consistent in both cases (Figures 2A and 3A). This outcome was similarly observed in the counting-cells assay measuring proliferation. The viability of cells with the non-tagged truncated mutant was also evaluated, revealing a significant difference compared to the wild-type cell line transfected with the mutant (Figure 3B). Consistently, the pR552* showed a high percentage of wound healing rate as compared to RB-HA (Figure 3C). In conclusion, we assert that the substantial effect observed with the mutation pR552* is attributed to the mutation itself and not an artefact of the HA-Tag.
The HApR552* mutant exhibits a gain of function behavior
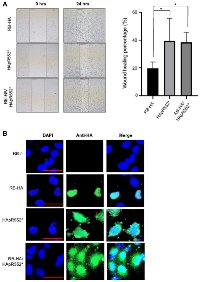
Subsequently, we aimed to investigate whether the truncated mutant, when expressed alongside a wild-type version of the protein, maintains this phenotype or if the presence of the wild-type RB protein rescues it. To test this, we co-transfected cells with both HApR552* and RB-HA and exposed the cells to mitomycin C treatment to inhibit proliferation. Subsequently, we induced a wound in the cell culture and measured the healing process to analyse migration. Our observations revealed that the co-transfected cells demonstrated a healing percentage similar to that of HApR552* alone. Both showed higher migration than the wild-type RB protein, as depicted in Figure 4A.
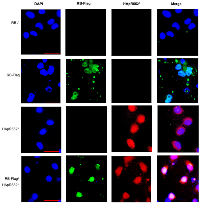
Furthermore, we conducted an immunofluorescence assay using the RB-HA wild type, the HApR552* mutant, and both in a co-transfected RB-HA and HApR552* experiment. We observed the classical nuclear localization pattern in the wild-type protein, whereas the truncated mutant exhibited nuclear-cytoplasmic localization. However, in the co-transfected cells, RB maintained the localization profile seen in the mutant alone, indicating that the wild-type protein does not rescue the aberrant behavior of HApR552* (Figure 4B).
Finally, we generated a construct with the wild-type tagged with a Flag-Tag, named RB-Flag to distinguish it from the wild-type HApR552*. Then, we performed an immunofluorescence assay co-transfected with both constructs and monitored the expression of RB-Flag in green and HApR552* in red. As observed in Figure 5, both constructs are expressed, and the pattern of both mutants is conserved. Surprisingly, even in the presence of the wild-type protein, the mutant allows the cells to maintain their carcinogenic characteristics, making this a gain-of-function mutant (Figures 4B, 5 and Supplementary Figure 3).
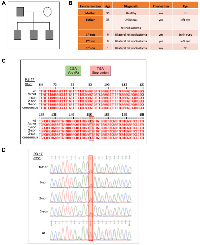
Mexican family carried the truncated mutant pR552*
The above results help us to explain the behavior shown by the Mexican family carrying the pR552* mutant named RBTR4 (Figure 6A). Residing in San Luis Potosí, México; this family comprises a mother, father, and three sons aged 9, 8, and 1 year (Figure 6B). The primers used to sequence the 27 exons of RB were based on the work of Mohd-Kahlid et al. 2015 [40]. The father was diagnosed with unilateral retinoblastoma at seven months old and enucleated from the left eye. He transmitted the mutant as an autosomal dominant trait with complete penetrance and high expressivity since the three sons showed bilateral retinoblastoma. The first son underwent enucleation of both eyes at one year old, while the second one was diagnosed at the age of 22 days and enucleated from the left eye at nine months. The third son is currently under treatment and conserved both eyes. The mother remains healthy. Peripheral venous blood was used for DNA extraction, followed by sequencing of the 27 exons. This sequencing revealed the presence of the pR552* truncated mutant in all male members of the family (Figure 6C, 6D). Taken together these results strongly indicate that the truncated mutant pR552* represents a gain-of-function mutation in the retinoblastoma protein, marking the first reported instance of such a mutation.
DISCUSSION
During the present work, we studied the phenotype of three different point mutants on the Rb1 gene observed in retinoblastoma patients: pD718N, pN328H, and pR552* with an HA-Tag. The three mutants exhibited distinct differences in proliferation, viability, migration, and localization. While two of them were localized in both the nucleus and cytoplasm, the third, even though localized only in the nucleus, was not observed in the nucleolus. Among the three mutants, only pR552* presented notable changes in viability experiments and a significant alteration in migration, with implications for its effects. As we continued analyzing the effects of this mutant, we prepared another cell line with the truncated mutant but without the HA-Tag to avoid potential interference. The results showed that, indeed, the new cell line behaves similarly to the previous one, indicating that the presence of the HA-Tag is not a factor influencing the previously observed results.
We wonder if a wild-type protein copy could reset the wild-type phenotype. The two-hit Knudson hypothesis establishes that most tumour suppressor genes require mutations in both alleles to develop cancer [18]. A single functional tumour suppressor gene copy is enough to withstand the perturbations of the mutated counterpart. For inherited cases, the first hit comes from one of the parents and is present in the other body cells, whereas the second hit is a somatic event in the patient’s retina. Surprisingly, the mutated phenotype persisted in co-transfected assays with a wild-type Rb1 gene, suggesting that just one hit is necessary to develop the disease.
A gain of function (GOF) mutation refers to the mutated gene product that has acquired a new molecular or biochemical function or even a new pattern of expression and is dominant or semi-dominant [41]. Many GOF mutations have been reported on different tumour suppressors, one of the most frequent being the transcription factor p53, the guardian of the genome [42, 43]. Some p53 missense mutations gain novel oncogenic activities [44–47]. However, patients carrying the mutated Rb1 gene somehow alter the structure of the Rb RNA, which can result in a truncated form of the protein that has been reported to be degraded. In any case, the mutation leads to a loss of function, with no reported gain-of-function (GOF) Rb1 mutants. The final consequence of this loss of function is continuous cell cycle progression, promoting dysregulated proliferation [37]. If this were true for the pR522* mutant, then it should not have any activity and cell proliferation parameters might be expected as the RB KO cell line. However, our results show that the pR552* truncated mutant protein is expressed; the resulting protein is truncated in the A pocket, with a size slightly more than half of the full-length RB. This portion of the protein is not only able to be expressed stably but also presents extended localization (nucleus and cytoplasm), exhibiting the highest proliferation when expressed in cells. In the migration assay, the truncated mutant pR552* also displays the highest activity compared with the wild-type RB protein and the other two mutations, pN328H and pD718N. We conclude that the pR552* mutant behaves as a GOF with a dominant phenotype (Figures 4 and 5).
These results contribute to explaining the behavior of retinal cancer in the RBTR4 family, where the truncated mutant exhibits complete penetrance and high expressivity. While the father showed a unilateral presentation, all three sons displayed bilateral diseases. Another surprising observation is that typically, if either the mother or father has a mutated gene, the child has a 50% chance of inheriting that mutated gene. However, in this family case, the father transmitted the mutant pR552* to 100% of his children.
MATERIALS AND METHODS
Patients
Patients involved in the study were part of the approved protocol, overseen by the research and ethics committee at the Central Hospital “Ignacio Morones Prieto”, San Luis Potosí, SLP, Mexico. Diagnostic and therapeutic manoeuvres were conducted following the Official Mexican Standard NOM-012-SSA3-2012, which outlines the criteria for executing research projects related to human health.
Inclusion criteria: Children with diagnosis of retinoblastoma, without a known diagnosis of other cancer. Exclusion criteria: Patients with diagnosis of other type of cancer. Controls: Healthy children at pediatric age and healthy adults in the same range of age of the family member patients.
Cell lines and mutants
The experiments were performed using the human H1299 cells (non-small lung carcinoma cell line) and H1299 RB−/− cells. The cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, L-glutamine 2 mM, penicillin 100 U/ml and streptomycin 100 μg/ml (Invitrogen). H1299 RB−/− was stably transfected by antibiotic selection with RB-HA wild-type, RBN328H, RBR552* and RBD718N. The full-length cDNA for human Rb was subcloned into the HindIII and BamHI sites of the pcDNA3.1 (RRID:Addgene_14743) vector with the previous addition of the label hemagglutinin (HA) tag in the C-terminal of RB. All mutants were generated by site-directed PCR mutagenesis using the previous Rb-HA template. Then RB-HA wild-type or mutants were released and subcloned in the pLNCX2 (RRID:Addgene_89818) retroviral vector with HindIII and NotI sites. Purified DNAs were used to produce retroviruses in HEK293T (RRID:CVCL_0063) cells by transiently transfecting the RB-HA wild-type or mutants together with Gag-pol (encodes for capsid proteins, reverse transcriptase and integrase) and VSV-G (encodes for the vesicular stomatitis virus G envelope protein) expressing plasmids, cells were transfected by lipofectamine 2000 reagent. Supernatants from transfected cells containing retroviral particles were collected 48 h after transfection and used to infect H1299-RB−/− cells in the presence of 8 μg/ml of polybrene for 8 h. 24h after infection cells were selected by adding G418 to the growth medium for at least 5 days. In addition, the full-length cDNA for human RB was subcloned into the HindIII and BamHI sites of the c-Flag pcDNA3.1 vector.
CRISPR/Cas9
The H1299 RB−/− cell line was built using the CRISPR Cas9 system. H1299 (RRID:CVCL_0060) cells were cotransfected with the RB CRISPR/Cas9 KO plasmid (Cat. No. sc-400116, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the RB HDR plasmid (Cat. No. sc-400116-HDR Santa Cruz Biotechnology, Santa Cruz, CA, USA). The RB CRISPR/Cas9 KO plasmid encodes for a guide RNA and nuclease, and the RB HDR plasmid encodes a homology-directed DNA repair template corresponding to the cutting sites generated by the RB CRISPR/Cas9 KO plasmid and a puromycin resistance gene to enable the selection of stable knockout cells. The single-celled clones were obtained by serial dilution and cultured with 3 μg/ml of puromycin (Cat. No. P7255, Sigma‐Aldrich). The clones were tested for RB expression through Western blot, and only the negatives for RB protein expression were amplified.
Western blot analysis
Each cell line was lysed using RIPA lysis buffer (200 mM NaCl, 0,2% NP-40, 10% (v/v) glycerol, 1 mM dithiothreitol (DTT), 1 mM EDTA and 25 mM Tris-HCl, pH 7,8) with a 1% protease inhibitor cocktail (Cat. No. 4693132001, Sigma‐Aldrich). Equal amounts of proteins were loaded into SDS-PAGE (polyacrylamide gel electrophoresis with sodium dodecyl sulfate) at 8%. The protein gels were transferred to a BioTrace NT nitrocellulose membrane (PALL Corporation) and blocked with 5% fat-free milk at PBS pH 7,4. The proteins were probed with the corresponding antibodies: anti-RB IF8 mouse (Cat. No. sc-102 Santa Cruz Biotechnology, CA, USA), anti-HA mouse (Cat. No. ab130275, Abcam, USA), and anti-actin-HRP (Cat. No. AC-15, Sigma-Aldrich).
RT-qPCR
The mRNA for each cell line was extracted using the TRIzol reagent. Reverse transcription to cDNA was performed using M-MuLV (Cat. No. N01-M0253S, New England Biolabs) reverse transcriptase with 1 μg of RNA. PCR amplification was performed in triplicate using the following primers: for human GAPDH, 5′-TCC AAA ATC AAG TGG GGC GA-3′ and 5′-TGA TGA CCC TTT TGG CTC CC-3′; for RB 5′CCT CTC GTC AGG CTT GAG TTT-3′ and 5′-GCT CTC TCT CTG ACA TGA TCT GG-3′. Each PCR mixture contained 1 μl of cDNA, 1ul of each primer (10 μM) and 5 μl of the SYBR Green (Thermofisher-K0221) in a final volume of 10 ul. The RT-qPCR was performed on 96-well microtiter plates using the 7500 Fast (Applied Biosystems) equipment. The amount of Rb mRNA (Ct of Rb) was normalized by subtracting the amount of GAPDH mRNA (GAPDH ct). The relative amount of Rb mRNA was obtained using the value 2−ΔΔCt.
Immunofluorescence
3 × 104 cells/well were seeded in a 24-well plate with a round coverslip for each cell line and incubated for 24 hours. Briefly, the cells were fixed with 3,7% paraformaldehyde for 30 min and permeabilized with PBS/Tritón X-100 (0.5%) for 1 h. Afterwards, the cells were washed with phosphate-buffered saline (PBS) for 5 min, followed by a 30 min incubation with PBS with bovine serum albumin (BSA) 3% to block. After blocking, the cells are incubated overnight at 4°C with 50 ul of the primary anti-HA mouse (Cat. No. ab130275, Abcam, USA) antibody at a dilution 1:100 prepared in PBS/BSA. Three washes of 5 min are done with 1X PBS and then incubated for 1 hour at 4°C with the secondary anti-mouse antibody conjugated with Alexa Fluor 568 (Cat. No. F0257, Sigma‐Aldrich) at a dilution of 1:1000 prepared in PBS/BSA solution. Afterwards, three washes of 5 min are made with 1X PBS at room temperature. Subsequently, the coverslips are assembled with 4 ul of the mounting solution containing 4′,6-diamidino-2-phenylindole (DAPI). The cells were photographed under a fluorescence microscope.
An additional experiment was conducted where 5 × 104 H1299 RB−/− cells were seeded per well in a 24-well plate with round coverslips and incubated for 24 hours. After incubation, they were co-transfected with RB-Flag and RB(R552*)-HA and incubated for 48 hours. Cells were fixed with 3.7% paraformaldehyde for 30 min and permeabilized with PBS/Tritón X-100 (0.5%) for 1 h. Afterwards, the cells were washed with PBS for 5 min, followed by a 30-minute incubation with PBS with 3% BSA to block. After blocking, cells are incubated overnight at 4°C with a mixture of the primary antibodies, anti-HA rabbit (Cat. No. ab236632, Abcam) at dilution of 1:100 and anti-Flag mouse (Cat. No. F1804, Sigma‐Aldrich) at dilution of 1:100 prepared in PBS/BSA. Three 5 min washes are performed with 1X PBS and then incubated for 1 hour at room temperature with a mixture of the secondary antibodies: anti-mouse antibody conjugated with Alexa Fluor 568 (Cat. No. F0257, Sigma‐Aldrich) 1:100 and anti-rabbit antibody conjugated with Rodamina (Cat. No. ab6718, Abcam, USA) 1:1000, prepared in PBS/BSA solution. Afterwards, three 5 min washes are performed with 1X PBS at room temperature. Subsequently, the coverslips are assembled with 4 μl of the mounting solution containing 4′,6-diamidino-2-phenylindole (DAPI). The cells were photographed under a fluorescence microscope.
MTT assay
1 × 104 cells/well were seeded in a 96-well plate in 100 ul of supplemented RPMI culture medium for each cell line and incubated for 48 hours. The MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was performed using the MTT Assay Kit (Cell Proliferation) (ab211091) according to the manufacturer’s protocol. Briefly, the MTT protocol used consisted on replacing the medium in which the cells are with a serum-free medium and MTT reagent (tetrazolium dye solution) and incubating them for 3 hours at 37ºC. After incubation, the cells were treated with MTT solvent for 15 minutes at room temperature. Absorbance was measured at OD = 595 nm.
Colony formation assay
Cells were cultured in a 6-well plate with a density of 200 cells/well density in 2 ml of supplemented RPMI culture medium and incubated for 10 days. The cells were then fixed with 100% methanol for 10 minutes of incubation at room temperature, and then the colonies formed were stained with 0.5% violet crystal solution for 10 minutes at room temperature. To remove the background stain, the cells were washed twice with ddH2O. Finally, the plaque was left to dry, and the colonies were photographed and counted with the “ColonyCount” application.
Wound-healing assay
7 × 104 cells/well were seeded in a 24-well plate in 500 ul of supplemented RPMI cultured medium and incubated for 24 hours. Cells were washed with 1X PBS and treated with mitomycin c (10 μg/ml for 1 hour) in a serum-free medium. After incubation, cells were again washed twice and left with 1% serum-supplemented medium. The cells were photographed at 0, 24 and 48 h to compare the migration capacity of the cell lines. Zen Lite 2012 microscopy and fluorescence software were used to analyse and quantify closure rates.
Statistics
Statistical analyses were performed using the GraphPad Prism 8.0 program (RRID:SCR_002798). Unidirectional analysis of variance (ANOVA) followed by the Bonferroni test was used to analyse the statistical differences between the groups. The results of the statistics were presented as a mean ± standard error of the mean (SEM). The p-value less than 0.05 was accepted as statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
Abbreviations
Rb1: Retinoblastoma gene; RB: Retinoblastoma protein; HApD718N: mutated protein D718N with a HA tag; HApR552*: mutated protein R552* with a HA tag; HApN328H: mutated protein N328H with a HA tag; pR552*, pN328H and pD718N: mutate proteins without a tag.
AUTHOR CONTRIBUTIONS
APB was responsible for performing the migration assay, the immunofluorescence experiments, forming the colony assay and MTT assay. MMS was responsible for the mutagenesis and the stable cell lines and for designing some protocols. IOS was responsible for doing the alpha-fold models and assisted with cell culture and proliferation experiments. KCG was responsible for PCR, sequencing the 27 exons of the patients, and data analysis. MMD was responsible for taking the peripherical blood samples of the patients, RNA extraction, and data analysis. MRCh was responsible for writing the research and ethics committee at the Central Hospital “Ignacio Morones Prieto”, as well as data analysis and discussion of the results. JHM was responsible for designing the experiments and performing western blots, analyzing data and giving feedback to the MS writing. VOI was responsible for designing the protocol and experiments, analyzing data, and writing MS and ethics protocol.
ACKNOWLEDGMENTS
We acknowledge the IQ. Celina Gonzalez Gallegos for technical support.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
ETHICAL STATEMENT
The study was developed within the framework of the approved protocol number 12–17 by the committee of research and ethics from the Central Hospital “Ignacio Morones Prieto” San Luis Potosí, SLP, Mexico. Diagnostic and therapeutic manoeuvres were carried out according to the Official Mexican Standard NOM-012-SSA3-2012, 2012 as well as with the current international codes for good practices in clinical research. The principles of the Helsinki Act of 1964 and its last revision in October 2013 were not transgressed.
CONSENT
The principles of the Helsinki Act of 1964, along with its last revision in October 2013, were adhered to, and written/signed informed consent was obtained from the guardians on behalf of the minors involved in the study.
FUNDING
This work was funded by CONAHCyT CF-2023-G-388. CONAHCyT CBF2023-2024-2470.
- 1. Retinoblastoma in the 20th century: past success and future challenges the Weisenfeld lecture. Invest Ophthalmol Vis Sci. 2005; 46:2683–91. https://doi.org/10.1167/iovs.04-1462. [Pubmed]
- 2. Epidemiological characteristics of retinoblastoma in children attending the Mexican Social Security Institute in Mexico City, 1990-94. Paediatr Perinat Epidemiol. 2002; 16:370–74. https://doi.org/10.1046/j.1365-3016.2002.t01-1-00442.x. [Pubmed]
- 3. International incidence of childhood cancer, 2001-10: a population-based registry study. Lancet Oncol. 2017; 18:719–31. https://doi.org/10.1016/S1470-2045(17)30186-9. [Pubmed]
- 4. Genetics of Retinoblastoma. Asia Pac J Ophthalmol (Phila). 2016; 5:260–64. https://doi.org/10.1097/APO.0000000000000219. [Pubmed]
- 5. Molecular alterations in retinoblastoma beyond RB1. Exp Eye Res. 2021; 211:108753. https://doi.org/10.1016/j.exer.2021.108753. [Pubmed]
- 6. Retinoblastoma: from discovery to clinical management. FEBS J. 2022; 289:4371–82. https://doi.org/10.1111/febs.16035. [Pubmed]
- 7. Analysis of the p53 pathway in peripheral blood of retinoblastoma patients; potential biomarkers. PLoS One. 2020; 15:e0234337. https://doi.org/10.1371/journal.pone.0234337. [Pubmed]
- 8. Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun Biol. 2020; 3:158. https://doi.org/10.1038/s42003-020-0873-9. [Pubmed]
- 9. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet. 2014; 51:366–74. https://doi.org/10.1136/jmedgenet-2014-102264. [Pubmed]
- 10. RB1 germline mutation spectrum and clinical features in patients with unilateral retinoblastomas. Ophthalmic Genet. 2021; 42:593–99. https://doi.org/10.1080/13816810.2021.1946703. [Pubmed]
- 11. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J Med Genet. 2014; 51:208–14. https://doi.org/10.1136/jmedgenet-2013-101821. [Pubmed]
- 12. RB1 gene mutations in retinoblastoma. Hum Mutat. 1999; 14:283–88. https://doi.org/10.1002/(SICI)1098-1004(199910)14:4<283::AID-HUMU2>3.0.CO;2-J. [Pubmed]
- 13. Non-synonymous, synonymous, and non-coding nucleotide variants contribute to recurrently altered biological processes during retinoblastoma progression. Genes Chromosomes Cancer. 2023; 62:275–89. https://doi.org/10.1002/gcc.23120. [Pubmed]
- 14. COSMIC: a curated database of somatic variants and clinical data for cancer. Nucleic Acids Research. 2024; 52: D1210–17. https://doi.org/10.1093/nar/gkad986.
- 15. Role of the RB-Interacting Proteins in Stem Cell Biology. Adv Cancer Res. 2016; 131:133–57. https://doi.org/10.1016/bs.acr.2016.04.002. [Pubmed]
- 16. RB1, development, and cancer. Curr Top Dev Biol. 2011; 94:129–69. https://doi.org/10.1016/B978-0-12-380916-2.00005-X. [Pubmed]
- 17. Retinoblastoma. Adv Exp Med Biol. 2010; 685:220–27. https://doi.org/10.1007/978-1-4419-6448-9_21. [Pubmed]
- 18. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971; 68:820–23. https://doi.org/10.1073/pnas.68.4.820. [Pubmed]
- 19. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020; 26:1549–56. https://doi.org/10.1038/s41591-020-1008-z. [Pubmed]
- 20. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am J Pathol. 2011; 179:1616–22. https://doi.org/10.1016/j.ajpath.2011.06.036. [Pubmed]
- 21. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013; 14:327–34. https://doi.org/10.1016/S1470-2045(13)70045-7. [Pubmed]
- 22. Comprehensive identification of pathogenic variants in retinoblastoma by long-and short-read sequencing. Cancer Lett. 2024; 598:217121. https://doi.org/10.1016/j.canlet.2024.217121. [Pubmed]
- 23. ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol Cell Biol. 2013; 33:3113–24. https://doi.org/10.1128/MCB.01597-12. [Pubmed]
- 24. Retinoblastoma protein partners. Adv Cancer Res. 2001; 82:1–54. https://doi.org/10.1016/s0065-230x(01)82001-7. [Pubmed]
- 25. Dual function of MDM2 and MDMX toward the tumor suppressors p53 and RB. Genes Cancer. 2016; 7:278–87. https://doi.org/10.18632/genesandcancer.120. [Pubmed]
- 26. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int J Mol Sci. 2017; 18:1776. https://doi.org/10.3390/ijms18081776. [Pubmed]
- 27. Mutant RB1 enhances therapeutic efficacy of PARPis in lung adenocarcinoma by triggering the cGAS/STING pathway. JCI Insight. 2023; 8:e165268. https://doi.org/10.1172/jci.insight.165268. [Pubmed]
- 28. Metastatic Competency and Tumor Spheroid Formation Are Independent Cell States Governed by RB in Lung Adenocarcinoma. Cancer Res Commun. 2023; 3:1992–2002. https://doi.org/10.1158/2767-9764.CRC-23-0172. [Pubmed]
- 29. Locally recurrent prostate cancer with RB1/TP53 alterations successfully treated by salvage focal brachytherapy: a case report. Am J Clin Exp Urol. 2023; 11:339–43. [Pubmed]
- 30. Case of Glioblastoma Multiforme in the Left Temporoparietal Region of the Brain. Cureus. 2022; 14:e28621. https://doi.org/10.7759/cureus.28621. [Pubmed]
- 31. The PyMOL molecular graphics system. in (Delano Scientific, S. C., Ca., ed). USA. 2004. http://pymol.sourceforge.net/.
- 32. Phosphorylation-dependent migration of retinoblastoma protein into the nucleolus triggered by binding to nucleophosmin/B23. Exp Cell Res. 2002; 276:233–41. https://doi.org/10.1006/excr.2002.5523. [Pubmed]
- 33. Clonogenic assay of cells in vitro. Nat Protoc. 2006; 1:2315–19. https://doi.org/10.1038/nprot.2006.339. [Pubmed]
- 34. In Vitro Tumorigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol Biol. 2018; 1692:89–95. https://doi.org/10.1007/978-1-4939-7401-6_8. [Pubmed]
- 35. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004; 18:2952–62. https://doi.org/10.1101/gad.322004. [Pubmed]
- 36. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996; 10:1621–32. https://doi.org/10.1101/gad.10.13.1621. [Pubmed]
- 37. Mutation spectrum of retinoblastoma patients in Vietnam. Mol Genet Genomic Med. 2023; 11:e2244. https://doi.org/10.1002/mgg3.2244. [Pubmed]
- 38. Characterization of the Portuguese population diagnosed with retinoblastoma. Sci Rep. 2022; 12:4378. https://doi.org/10.1038/s41598-022-08326-6. [Pubmed]
- 39. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003; 72:253–69. https://doi.org/10.1086/345651. [Pubmed]
- 40. Spectrum of germ-line RB1 gene mutations in Malaysian patients with retinoblastoma. Mol Vis. 2015; 21:1185–90. [Pubmed]
- 41. Genetic mapping and manipulation: chapter 8--Dominant mutations. WormBook. 2006; 1–6. https://doi.org/10.1895/wormbook.1.97.1. [Pubmed]
- 42. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007; 9:573–80. https://doi.org/10.1038/ncb1571. [Pubmed]
- 43. Gain of function of p53 cancer mutants in disrupting critical DNA damage response pathways. Cell Cycle. 2007; 6:1570–73. https://doi.org/10.4161/cc.6.13.4456. [Pubmed]
- 44. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 2001; 20:4163–72. https://doi.org/10.1093/emboj/20.15.4163. [Pubmed]
- 45. The C-terminus of mutant p53 is necessary for its ability to interfere with growth arrest or apoptosis. Oncogene. 2001; 20:4891–98. https://doi.org/10.1038/sj.onc.1204724. [Pubmed]
- 46. Mutant p53 gain-of-function stimulates canonical Wnt signaling via PI3K/AKT pathway in colon cancer. J Cell Commun Signal. 2023; 17:1389–403. https://doi.org/10.1007/s12079-023-00793-4. [Pubmed]
- 47. p53 isoforms gain functions. Oncogene. 2010; 29:5113–19. https://doi.org/10.1038/onc.2010.266. [Pubmed]
 All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.